Souhrnné informace o léku - ZOLEDRONIC ACID FRESENIUS KABI
1. NÁZEV PŘÍPRAVKU
Zoledronic acid Fresenius Kabi 4 mg/5 ml koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOTENÍ
Jedna injekční lahvička s 5 ml koncentrátu obsahuje acidum zoledronicum 4 mg (ve formě acidum zoledronicum monohydricum)
Jeden ml koncentrátu obsahuje acidum zoledronicum 0,8 mg (ve formě acidum zoledronicum monohydricum).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok (sterilní koncentrát)
Čirý bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
– Prevence kostních příhod (patologických zlomenin, kompresivních zlomenin obratlů, radiační nebo chirurgická léčba kostí nebo nádorem indukovaná hyperkalcemie) u dospělých pacientů s pokročilým maligním onemocněním postihujícím kosti.
– Léčba dospělých pacientů s hyperkalcemií vyvolanou nádorovým onemocněním (TIH).
4.2 Dávkování a způsob podání
Přípravek Zoledronic acid Fresenius Kabi musí být předepisován a podáván pacientům pouze zdravotnickými pracovníky se zkušenostmi s intravenózním podáváním bisfosfonátů. Pacienti léčeni přípravkem Zoledronic acid Fresenius Kabi mají dostat příbalovou informaci a informační kartu pacienta.
Dávkování
Prevence kostních příhod u pacientů s pokročilým maligním onemocněním postihujícím kosti
Dospělí a starší lidé
Doporučená dávka k prevenci kostních příhod u pacientů s pokročilým nádorovým onemocněním postihujícím kosti je 4 mg kyseliny zoledronové každé 3 až 4 týdny.
Pacientům má být také perorálně podán denně doplněk 500 mg kalcia a 400 IU vitaminu D.
Při rozhodování, zda léčit pacienty s kostními metastázami za účelem prevence kostních příhod je nutno vzít v úvahu, že se účinky léčby projeví za 2–3 měsíce.
Léčba TIH
Dospělí a starší lidé
Doporučená dávka při léčbě hyperkalcemie (kalcium v séru korigované na albumin > 12,0 mg/dl nebo 3,0 mmol/l) je 4 mg kyseliny zoledronové v jedné dávce.
Zhoršená funkce ledvin
TIH:
U pacientů s TIH trpících současně závažným zhoršením funkce ledvin se smí o léčbě přípravkem Zoledronic acid Fresenius Kabi uvažovat až po zhodnocení rizika a přínosu léčby. Pacienti s hladinou kreatininu v séru > 400 |imoi/i nebo > 4,5 mg/dl byli z klinických studií vyloučeni. U pacientů s TIH se sérovým kreatininem < 400 gmol/l nebo < 4,5 mg/dl není nutná úprava dávkování (viz bod 4.4).
Prevence kostních příhod u pacientů s pokročilou formou maligního onemocnění postihujícího kosti:
U pacientů s mnohočetným myelomem nebo metastázami solidních nádorů do kostí musí být při zahájení léčby přípravkem Zoledronic acid Fresenius Kabi stanoven sérový kreatinin a clearance kreatininu (CLcr). CLcr se vypočte ze sérového kreatininu pomocí Cockcroft-Gaultova vzorce. Zoledronic acid Fresenius Kabi se nedoporučuje podávat pacientům, kteří již před zahájením léčby trpí závažným poškozením funkce ledvin, které je u této populace definováno jako CLcr < 30 ml/min. Pacienti s hladinou kreatininu v séru > 265 ^mol/l nebo > 3,0 mg/dl byli z klinických studií vyloučeni.
U pacientů s kostními metastázami a s mírným nebo středním postižením funkce ledvin před zahájením léčby, které bylo pro tuto skupinu pacientů definováno jako CLcr 30 – 60 ml/min, se doporučuje následující dávkování přípravku Zoledronic acid Fresenius Kabi (viz bod 4.4):
| Výchozí hodnoty clearance kreatininu (ml/min) | Doporučené dávkování Zoledronic acid Fresenius Kabi* |
| > 60 | 4,0 mg kyseliny zoledronové |
| 50–60 | 3,5 mg* kyseliny zoledronové |
| 40–49 | 3,3 mg* kyseliny zoledronové |
| 30–39 | 3,0 mg* kyseliny zoledronové |
* Dávky byly vypočteny z předpokládané AUC 0,66 (mgJiod/l) (CLcr = 75 ml/min). Při podávání snížených dávek pacientům se zhoršenou funkcí ledvin je možné očekávat dosažení stejných AUC, jaké byly pozorovány u pacientů s clearance kreatininu 75 ml/min.
Po zahájení léčby musí být stanovován sérový kreatinin před podáním každé dávky přípravku Zoledronic acid Fresenius Kabi a při zhoršení renálních funkcí musí být léčba přerušena. V klinických studiích bylo zhoršení funkce ledvin definováno následovně:
-
– U pacientů s normální výchozí hodnotou sérového kreatininu (< 1,4 mg/dl nebo < 124 ^mol/l), zvýšení o 0,5 mg/dl nebo 44 ^mol/l;
-
– U pacientů s abnormálním výchozími hodnotami sérového kreatininu (> 1,4 mg/dl nebo > 124 ^mol/l), zvýšení o 1,0 mg/dl nebo 88 ^mol/l.
V klinických studiích bylo podávání kyseliny zoledronové znovu zahájeno pouze tehdy, pokud se hodnota kreatininu vrátila do rozmezí 10 % od výchozí hodnoty (viz bod 4.4). Léčba přípravkem Zoledronic acid Fresenius Kabi má být obnovena stejnou dávkou, která byla podávána před přerušením.
Pediatrická populace
Bezpečnost a účinnost kyseliny zoledronové u dětí ve věku 1 rok až 17 let nebyla stanovena. V současnosti dostupné údaje jsou popsány v bodech 5.1 a 5.2, ale na jejich základě nelze doporučit dávkování u dětí.
Způsob podání
Intravenózní podání
Přípravek Zoledronic acid Fresenius Kabi 4 mg/5 ml, koncentrát pro infuzní roztok, následně naředěný ve 100 ml (viz bod 6.6), by měl být podáván jako jednorázová intravenózní infuze po dobu alespoň 20 minut.
U pacientů s mírnou až středně těžkou poruchou funkce ledvin jsou doporučené redukované dávky přípravku Zoledronic acid Fresenius Kabi (viz bod “Dávkování” výše a bod 4.4).
Instrukce pro přípravu redukovaných dávek přípravku Zoledronic acid Fresenius Kabi
Odeberte odpovídající objem koncentrátu pro infuzní roztok:
-
– 4,4 ml pro dávku 3,5 mg
-
– 4,1 ml pro dávku 3,3 mg
-
– 3,8 ml pro dávku 3,0 mg
Návod k naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
Odebrané množství koncentrátu pro infuzní roztok musí být dále naředěno ve 100 ml sterilního roztoku 0,9% chloridu sodného nebo v roztoku 5% glukózy. Dávka musí být podána v jedné intravenózní infuzi po dobu nejméně 20 minut.
Zoledronic acid Fresenius Kabi, koncentrát, nesmí být mísen s infuzními roztoky obsahujícími kalcium nebo jiné bivalentní kationty jako je laktátový Ringerův roztok a musí být podáván odděleně jako samostatný intravenózní roztok oddělenou infuzní linkou.
Před a po podání přípravku Zoledronic acid Fresenius Kabi musí být pacienti dobře hydratováni.
4.3 Kontraindikace
4.4 Zvláštní upozornění a opatření pro použití
Obecná
Před podáním přípravku Zoledronic acid Fresenius Kabi musí být posouzen stav hydratace pacientů, a v případě potřeby musí být hydratace adekvátně upravena.
U pacientů s rizikem srdečního selhání je nutné zabránit nadměrnému přívodu tekutin.
Po zahájení terapie přípravkem Zoledronic acid Fresenius Kabi musí být pečlivě sledovány standardní metabolické parametry související s hyperkalcemií, jako jsou sérové hladiny kalcia, fosfátu a magnesia. Pokud dojde ke vzniku hypokalcemie, hypofosfatemie nebo hypomagnesemie, je nutné zahájit krátkodobou doplňkovou léčbu. Pacienti s neléčenou hyperkalcemií mají obvykle do jisté míry poškozenou funkci ledvin, proto je nutné uvažovat o pečlivém sledování ledvinných funkcí.
Jiné léčivé přípravky obsahující kyselinu zoledronovou jako léčivou látku bývají indikovány při osteoporóze a při léčbě Pagetovy kostní choroby. Pacienti léčení přípravkem Zoledronic acid Fresenius Kabi nemají být současně léčeni jinými léčivými přípravky obsahujícími kyselinu zoledronovou nebo jinými bisfosfonáty, protože kombinované účinky těchto látek nejsou známy.
Ledvinná nedostatečnost
U pacientů s TIH a zjištěným zhoršováním funkcí ledvin musí být velmi dobře posouzen jejich stav a zváženo, zda přínos léčby přípravkem Zoledronic acid Fresenius Kabi převáží možné riziko léčby.
Při rozhodnutí léčit pacienty s kostními metastázami pro prevenci kostních příhod by se mělo vzít v úvahu, že nástup léčebného účinku je za 2 až 3 měsíce.
Používání kyseliny zoledronové v souladu s indikacemi v bodech 4.1 a 4.2 bylo spojeno s hlášením poruch funkce ledvin. Mezi faktory, které mohou zvyšovat riziko zhoršení ledvinných funkcí patří dehydratace a již existující poškození funkce ledvin, opakované cykly podávání přípravku Zoledronic acid Fresenius Kabi nebo jiných bisfosfonátů, stejně jako jiných nefrotoxických léčivých přípravků. Vznik renálního poškození v souvislosti s podáváním kyseliny zoledronové může souviset s vysokou maximální plasmatickou koncentrací zvyšující intracelulární koncentraci kyseliny zoledronové a tím riziko poškození buněk. Přestože je toto riziko sníženo při podávání kyseliny zoledronové v dávce 4 mg po dobu delší než 20 minut, může se poškození ledvin přesto vyskytnout. Bylo hlášeno zhoršení ledvinných funkcí a progrese do ledvinného selhání s nutností dialýzy u pacientů po úvodní nebo jednorázové dávce 4 mg kyseliny zoledronové. Zvýšení kreatininu v séru se může u některých pacientů vyskytnout po opakovaném podání přípravku Zoledronic acid Fresenius Kabi v dávkách doporučených k prevenci kostních příhod, i když méně často.
Před každou aplikací přípravku Zoledronic acid Fresenius Kabi musí být pacientům stanovena hladina sérového kreatininu. U pacientů s kostními metastázami a s mírným až středně závažným poškozením funkce ledvin se doporučuje zahájit léčbu nižšími dávkami přípravku Zoledronic acid Fresenius Kabi. U pacientů, u kterých bylo v průběhu léčby přípravkem Zoledronic acid Fresenius Kabi prokázáno zhoršení funkce ledvin, musí být léčba přípravkem Zoledronic acid Fresenius Kabi přerušena do doby, než se hladina sérového kreatininu vrátí na hodnoty, které se nebudou lišit o více než 10 % od výchozí hodnoty.
Léčba přípravkem Zoledronic acid Fresenius Kabi má pokračovat stejnou dávkou, jaká byla podávaná před přerušením léčby.
Vzhledem k možnému vlivu kyseliny zoledronové na funkci ledvin se pro nedostatek klinických údajů o bezpečnosti podávání u pacientů se závažným poškozením funkce ledvin již před zahájením léčby (v klinických studiích definovaných hladinou kreatininu v séru > 400 ^mol/l nebo > 4,5 mg/dl u pacientů s TIH a > 265 gmol/l nebo > 3,0 mg/dl u pacientů s karcinomem a kostními metastázami) a z důvodu pouze omezeného množství farmakokinetických údajů u těchto pacientů (clearance kreatininu < 30 ml/min) podávání přípravku Zoledronic acid Fresenius Kabi pacientům se závažným poškozením funkce ledvin nedoporučuje.
Jaterní nedostatečnost
U pacientů se závažnou jaterní insuficiencí je k dispozici jen omezené množství klinických údajů, a proto nemohlo být dáno specifické doporučení pro tuto skupinu pacientů.
Osteonekróza
Osteonekróza čelisti
U pacientů používajících kyselinu zoledronovou byly v klinických studiích a po uvedení přípravku na trh méně často hlášeny případy osteonekrózy čelisti (OČ).
U pacientů s nehojícími se lézemi měkkých tkání v ústech by mělo být s výjimkou akutních medicínských stavů zahájení léčby nebo nového cyklu léčby odloženo.
Před zahájením léčby bisfosfonáty je u pacientů s konkomitantními rizikovými faktory doporučené zubní vyšetření s případným preventivním ošetřením a individuálním vyhodnocením poměru prospěchu-rizika.
Při vyhodnocení individuálního rizika vzniku OČ mají být zvažovány následující rizikové faktory:
-
– Účinnost bisfosfonátů (vyšší riziko pro vysoce účinné látky), cesta podání (vyšší riziko pro parenterální podání) a kumulativní dávka bisfosfonátů
-
– Maligní nádorové onemocnění, komorbidity (např. anémie, koagulopatie, infekce), kouření
-
– Konkomitantní terapie: chemoterapie, inhibitory angiogeneze (viz bod 4.5), radioterapie krku a hlavy, kortikosteroidy
-
– Stomatologická onemocnění v anamnéze, špatná ústní hygiena, periodontální onemocnění, invazivní stomatologické zákroky (např. extrakce zubů) a špatně naléhající zubní protézy
Všichni pacienti mají být vyzváni, aby během léčby přípravkem Zoledronic acid Fresenius Kabi udržovali dobrou ústní hygienu, absolvovali rutinní vyšetření chrupu a okamžitě hlásili jakékoli příznaky v ústech, jako je kývání zubů, bolest nebo otoky nebo nehojící se léze nebo výtok. Během léčby mají být invazivní stomatologické procedury prováděny pouze po pečlivém vyhodnocení a nemají se provádět v době blízké podání kyseliny zoledronové.
U pacientů, kde se osteonekróza čelisti vyvine během bisfosfonátové léčby, může stomatologický výkon zhoršit stav. Neexistují data, která by dokládala, že vysazení bisfosfonátové léčby snižuje riziko osteonekrózy čelisti u pacientů vyžadujících stomatologický výkon.
Plán léčby pacientů s OČ by měl být navržen v úzké spolupráci mezi ošetřujícím lékařem a dentistou nebo zubním chirurgem s odbornou znalostí OČ. Dokud se stav nezlepší a pokud možno nevymizí přispívající rizikové faktory, mělo by být zváženo dočasné přerušení léčby kyselinou zoledronovou.
Osteonekróza lokalizovaná v jiných částech těla
-
V souvislosti s léčbou bisfosfonáty byla hlášena osteonekróza zevního zvukovodu, zejména při dlouhodobém podávání. Mezi možné rizikové faktory osteonekrózy zvukovodu patří používání steroidů a chemoterapie a/nebo lokální rizikové faktory, jako například infekce nebo trauma. Možnost vzniku osteonekrózy zevního zvukovodu je třeba zvážit u pacientů léčených bisfosfonáty, kteří mají ušní symptomy včetně chronických infekcí ucha. Navíc došlo ke sporadickým hlášením osteonekrózy jiných částí skeletu, zahrnující osteonekrózu kyčle a stehenní kosti, hlášených převážně u dospělých pacientů s nádorem léčených přípravkem Zoledronic acid Fresenius Kabi.
Bolesti muskuloskeletálního systému
Z postmarketingových zkušeností byly hlášeny silné bolesti kostí, kloubů a/nebo svalů občas zneschopňující pacienty, kterým byla podána kyselina zoledronová v souladu s indikacemi v bodech 4.1 a 4.2. Nicméně tato hlášení byla řídká. Doba do počátku nástupu projevů nežádoucích účinků od zahájení léčby je rozličná, od jednoho dne až po několik měsíců. Jakmile byla léčba přerušena, většině pacientů se od příznaků ulevilo. Podskupina pacientů zaznamenala opětovný návrat symptomů poté, co byla znovu léčena kyselinou zoledronovou nebo jiným bisfosfonátem.
Atypické zlomeniny femuru
-
V souvislosti s léčbou bisfosfonáty byly hlášeny atypické subtrochanterické a diafyzární zlomeniny femuru, zejména u pacientů dlouhodobě léčených pro osteoporózu. Tyto příčné nebo krátké šikmé zlomeniny se mohou objevit kdekoli v celé délce femuru od oblasti těsně pod malým trochanterem až do části těsně nad suprakondylickým rozšířením. Tyto zlomeniny se objevují po minimálním traumatu nebo bez souvislosti s ním a u některých pacientů se mohou projevovat bolestí ve stehně nebo třísle, často sdružené na zobrazovacích vyšetřeních s obrazem typickým pro stresové zlomeniny (neobvyklé nízkotraumatické zlomeniny, v angličtině známé jako „insufficiency fractures“), týdny až měsíce před manifestací kompletní zlomeniny femuru. Zlomeniny jsou často oboustranné, proto je nutné u pacientů léčených bisfosfonáty, kteří utrpěli zlomeninu diafýzy femuru, vyšetřit i kontralaterální femur. Rovněž bylo zaznamenáno špatné hojení těchto zlomenin. U pacientů, u kterých je podezření na atypickou zlomeninu femuru, je třeba při hodnocení jejich stavu zvážit i přerušení léčby bisfosfonáty, a to na základě zhodnocení prospěchu a rizika léčby u jednotlivého pacienta.
Pacienty je třeba poučit, aby během léčby bisfosfonáty hlásili jakoukoli bolest v oblasti stehna, kyčle nebo třísla, a všechny pacienty, u kterých se tyto příznaky objeví, je třeba vyšetřit s ohledem na možnou inkompletní zlomeninu femuru.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v dávce, je tedy v podstatě „bez sodíku“.
Hypokalcémie
U pacientů léčených kyselinou zoledronovou byla hlášena hypokalcémie. V důsledku těžké hypokalcémie byly hlášeny srdeční arytmie a neurologické nežádoucí účinky (zahrnující křeče, hypoestézii a tetanii). Byly hlášeny případy závažné hypokalcemie vyžadující hospitalizaci. V některých případech může být hypokalcemie život ohrožující (viz bod 4.8). Při podávání přípravku Zoledronic acid Fresenius Kabi spolu s léčivými přípravky, které mohou způsobit hypokalcémii, je nutné dbát opatrnosti, protože mohou mít synergní účinek vyúsťující v závažnou hypokalcémii (viz bod 4.5). Před zahájením léčby přípravkem Zoledronic acid Fresenius Kabi má být změřena hladina vápníku v séru a upravena hypokalcémie. Pacientům má být dodáváno přiměřené množství vápníku a vitamínu D.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakcí
V klinických studiích byla kyselina zoledronová, používaná v souladu s indikacemi v bodech 4.1 a 4.2, podávána souběžně s běžně používanými protinádorovými léky, diuretiky, antibiotiky a analgetiky, aniž by byl pozorován výskyt klinicky zřejmých interakcí. Při studiu in vitro (viz bod 5.2) nebyla patrná vazba kyseliny zoledronové na plazmatické bílkoviny, ani nebyla zjištěna inhibice lidských P450 enzymů, ale žádná klinická studie interakcí nebyla provedena.
Při souběžné aplikaci bisfosfonátů s aminoglykosidy, kalcitoninem nebo kličkovými (loop) diuretiky se doporučuje zvláštní opatrnost, protože může dojít k aditivnímu účinku těchto léků, s následným snížením hladiny kalcia v séru na delší dobu, než je požadováno (viz bod 4.4).
Opatrnost je také nutná, pokud je přípravek Zoledronic acid Fresenius Kabi indikován společně s jinými potenciálně nefrotoxickými léčivými přípravky. Je také nutné věnovat zvýšenou pozornost možnému vývoji hypomagnesemie během léčby.
U pacientů s mnohočetným myelomem může být zvýšené riziko poškození ledvin, pokud je přípravek Zoledronic acid Fresenius Kabi podávan v kombinaci s thalidomidem.
Je nutná opatrnost, pokud je přípravek Zoledronic acid Fresenius Kabi podáván s antiangiogenními léčivými přípravky, protože u pacientů léčených současně těmito léčivými přípravky byl hlášen zvýšený výskyt osteonekrózy čelisti.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Adekvátní údaje o podávání kyseliny zoledronové těhotným ženám nejsou k dispozici. Reprodukční studie provedené s kyselinou zoledronovou na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známé. Přípravek Zoledronic acid Fresenius Kabi nemá být během těhotenství podáván. Ženám ve fertilním věku má být doporučeno vyhnout se otěhotnění.
Kojení
Není známo, zda je kyselina zoledronová vylučována do mateřského mléka. Přípravek Zoledronic acid Fresenius Kabi je u kojících matek kontraindikován (viz bod 4.3).
Fertilita
Potenciální nežádoucí účinky kyseliny zoledronové na fertilitu v parentální a F1 generaci byly hodnoceny u potkanů. Došlo k výrazným farmakologickým účinkům, které je možné považovat za součást inhibice metabolizace vápníku v kostech, což se projevilo peripartální hypokalcemií (skupinovým účinkem bisfosfonátů), dystokií a časným ukončením studie. Z tohoto důvodu nemohl být určen konečný vliv kyseliny zoledronové na fertilitu lidí.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nežádoucí účinky, jako je závrať a ospalost, mohou mít vliv na schopnost řídit nebo obsluhovat stroje, proto je třeba dbát zvýšené opatrnosti při řízení a obsluze strojů během používání přípravku Zoledronic acid Fresenius Kabi.
4.8 Nežádoucí účinky
Přehled bezpečnostního profilu
Během tří dnů po podání kyseliny zoledronové, podané v souladu s indikacemi v bodech 4.1 a 4.2, byly často hlášeny reakce akutní fáze s příznaky zahrnujícími bolest kostí, horečku, únavu, artralgii, myalgii, ztuhlost a artritidu s následnými otoky kloubů; tyto příznaky obvykle ustupují během několika dní (viz popis vybraných nežádoucích účinků).
Následují významná identifikovaná rizika kyseliny zoledronové ve schválených indikacích:
Porucha funkce ledvin, osteonekróza čelisti, reakce akutní fáze, hypokalcemie, fibrilace síní, anafylaxe, intersticiální onemocnění plic. Frekvence pro každé z těchto identifikovaných rizik jsou uvedeny v Tabulce 1.
Seznam nežádoucích účinků v tabulce
Následující nežádoucí účinky, uvedené níže v tabulce 1, byly získány z klinických studií a postmarketingového sledování převážně při dlouhodobé léčbě 4 mg kyseliny zoledronové:
Podle frekvence výskytu byly nežádoucí účinky řazeny od nejčastěji se vyskytujících pomocí následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
| Poruchy krve a lymfatického systému | |
| Časté: | Anemie |
| Méně časté: | Trombocytopenie, leukopenie |
| Vzácné: | Pancytopenie |
| Poruchy imunitního systému | |
| Méně časté: | Hypersenzitivní reakce |
| Vzácné: | Angioneurotický edém |
| Psychiatrické poruchy | |
| Méně časté: | Úzkost, poruchy spánku |
| Vzácné: | Zmatenost |
| Poruchy nervového systému | |
| Časté: | Bolesti hlavy |
| Méně časté: | Závratě, parestezie, dysgeuzie, hypoestézie, |
| hyperestézie, tremor, ospalost | |
| Velmi vzácné: | Křeče, hypoestézie a tetanie (v důsledku |
| hypokalcemie) | |
| Poruchy oka | |
| Časté: | Konjunktivitida |
| Méně časté: | Rozmazané vidění, skleritida a zánět očnice |
| Vzácné: | Uveitida |
| Velmi vzácné: | Episkleritida |
| Srdeční poruchy | |
| Méně časté: | Hypertenze, hypotenze, fibrilace síní, hypotenze vedoucí k synkopě nebo oběhovému kolapsu |
| Vzácné: | Bradykardie, srdeční arytmie (v důsledku hypokalcémie) |
| Respirační, hrudní a mediastinálníporuchy | |
| Méně časté: | Dušnost, kašel, bronchokonstrikce |
| Vzácné: | Intersticiální onemocnění plic |
| Gastrointestinální poruchy | |
| Časté: | Nauzea, zvracení, snížená chuť k jídlu |
| Méně časté: | Průjem, zácpa, bolesti břicha, dyspepsie, stomatitida, sucho v ústech |
| Poruchy kůže a podkožní tkáně | |
| Méně časté: | Svědění, vyrážka (včetně erytematózní a makulární vyrážky), zvýšené pocení |
| Poruchy svalové a kosterní soustavy a pojivové tkáně | |
| Časté: | Bolestivost kostí, svalů a kloubů, generalizovaná bolest |
| Méně časté: | Svalové křeče, osteonekróza čelisti |
| Velmi vzácné: | Osteonekróza zevního zvukovodu (skupinový nežádoucí účinek bisfosfonátů) a jiných částí skeletu zahrnující osteonekrózu kyčle a stehenní kosti |
| Poruchy ledvin a močových cest | |
| Časté: | Zhoršení ledvinných funkcí |
| Méně časté: | Akutní selhání ledvin, hematurie, proteinurie |
| Vzácné: | Získaný Fanconiho syndrom |
| Celkové poruchy a reakce v místě aplikace | |
| Časté: | Horečka, chřipce podobné příznaky (včetně únavy, třesavky, malátnosti a návalů horka) |
| Méně časté: | Astenie, periferní otoky, reakce v místě vpichu (včetně bolesti, podráždění, otoku, zatuhnutí), bolestivost na hrudi, zvýšení tělesné hmotnosti, anafylaktická reakce/šok, kopřivka |
| Vzácné: | Artritida a otoky kloubů jako příznak reakce akutní fáze |
| Vyšetření | |
| Velmi časté: | Hypofosfatemie |
| Časté: | Zvýšení kreatininu a urey v krvi, hypokalcemie |
| Méně časté: | Hypomagnezemie, hypokalemie |
| Vzácné: | Hyperkalemie, hypernatremie |
Popis vybraných nežádoucích účinků
Porucha _ funkce ledvin
Léčba kyselinou zoledronovou, podanou v souladu s indikacemi v bodech 4.1 a 4.2, byla spojena s hlášením renální dysfunkce. V poolované analýze bezpečnostních dat získaných v registračních studiích kyseliny zoledronové podávané k předcházení kostních příhod u pacientů s formou pokročilého maligního onemocnění postihujícího kosti, byly četnosti poruch nežádoucích účinků spojených s poruchou funkce ledvin vzniklé v možné souvislosti s podáváním kyseliny zoledronové (nežádoucí reakce) následující: mnohočetný myelom (3,2%), karcinom prostaty (3,1%), karcinom prsu (4,3%), karcinom plic a další solidní nádory (3,2%). Faktory, které mohou zvýšit potenciál ke zhoršení funkce ledvin, zahrnují dehydrataci, již existující poškození ledvin, mnohočetné cykly kyseliny zoledronové nebo jiných bisfosfonátů, stejně jako souběžné užívání nefrotoxických léčivých přípravků nebo použití kratší doby infuze, než je doporučeno. Zhoršení funkce ledvin, progrese k renálnímu selhání a dialýze byly hlášeny u pacientů po počáteční dávce nebo jednotlivé dávce 4 mg kyseliny zoledronové (viz bod 4.4).
Osteonekróza čelisti
Byly popsány případy osteonekrózy čelisti převážně u onkologických pacientů léčených léčivými přípravky inhibujícími kostní resorpci, jako je přípravek Zoledronic Acid Fresenius Kabi (viz bod 4.4). Mnoho těchto pacientů také dostávalo chemoterapii a kortikosteroidy a mělo příznaky lokální infekce včetně osteomyelitidy. Většina hlášení se týkala onkologických pacientů po extrakci zubu nebo jiném stomatochirurgickém výkonu.
Fibrilace síní
V jedné 3leté, randomizované, dvojitě zaslepené kontrolované studii, která hodnotila účinnost a bezpečnost kyseliny zoledronové 5 mg podávané jednou ročně oproti placebu při léčbě postmenopauzální osteoporózy (PMO), byl celkový výskyt fibrilace síní 2,5 % (96 z 3862) u pacientů používajících kyselinu zoledronovou 5 mg a 1,9 % (75 z 3852) u pacientů s placebem. Poměr vážných případů fibrilace síní byl 1,3 % (51 z 3862) u pacientů používajících kyselinu zoledronovou 5 mg a 0,6 % (22 z 3852) u pacientů používajících placebo. Tento nežádoucí účinek pozorovaný ve studii nebyl zaznamenán v jiných studiích s kyselinou zoledronovou, včetně studií s kyselinou zoledronovou 4 mg podávanou každé 3–4 týdny onkologickým pacientům. Mechanismus původu zvýšeného výskytu fibrilace síní v této jediné klinické studii není známý.
Reakce akutní fáze
Tento nežádoucí účinek se skládá ze souboru příznaků, které zahrnují horečku, myalgii, bolest hlavy, bolest končetin, nauzeu, zvracení, průjem, artralgii a artritidu s následnými otoky kloubů. Nastupuje < 3 dny po infuzi kyseliny zoledronové (podané v souladu s indikacemi v bodech 4.1 a 4.2) a reakce je také známá pod názvy “flu-like syndrom” (syndrom podobný chřipce) nebo “post-dose” syndrom (příznaky po podání dávky).
Atypické zlomeniny femuru
Po uvedení přípravku na trh byly hlášeny následující nežádoucí účinky (frekvence vzácná):
Atypické subtrochanterické a diafyzární zlomeniny femuru (skupinový nežádoucí účinek bisfosfonátů).
Nežádoucí účinky spojené s hypokalcemií
Hypokalcemie je důležitým známým rizikem podání kyseliny zoledronové ve schválených indikacích. Na základě hodnocení případů z klinických studií a případů po uvedení na trh existují dostatečné důkazy pro podporu souvislosti mezi léčbou kyselinou zoledronovou, hlášenými případy hypokalcemie a následným výskytem srdeční arytmie. Dále existují důkazy o souvislosti mezi hypokalcemií a následnými neurologickými nežádoucími účinky hlášenými v těchto případech, které zahrnují křeče, hypoestézii a tetanii (viz bod 4.4).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
Klinické zkušenosti s akutním předávkováním kyselinou zoledronovou jsou omezené. Bylo hlášeno chybné podávání kyseliny zoledronové v dávkách až do 48 mg. Pacienti, kteří dostávali vyšší dávky, než je dávka doporučená (viz bod 4.2), musí být pečlivě sledováni, protože bylo pozorováno poškození renálních funkcí (včetně renálního selhání) a odchylky v hladinách sérových koncentrací elektrolytů (včetně vápníku, fosforu a hořčíku). V případě hypokalcemie se dle klinické indikace může podat infuze kalcium-glukonátu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii nemocí kostí, bisfosfonáty, ATC kód: M05BA08
Zoledronová kyselina patří do skupiny bisfosfonátů, které působí primárně v kostech. Je to inhibitor osteoklastické resorpce kostí.
Selektivní působení bisfosfonátů v kostech spočívá v jejich vysoké afinitě k mineralizované kosti, ale přesný mechanizmus účinku vedoucího k inhibici osteoklastické aktivity zůstává stále neobjasněn. V dlouhodobých studiích na zvířatech inhibovala kyselina zoledronová kostní resorpci bez nežádoucího ovlivnění tvorby, mineralizace nebo mechanických vlastností kostí.
Kromě inhibice kostní resorpce má kyselina zoledronová navíc některé protinádorové vlastnosti, které by mohly přispívat k celkové účinnosti léčby kostních metastáz. V preklinických studiích byly demonstrovány následující vlastnosti:
-
– In vivo: inhibice osteoklastické kostní resorpce, která ovlivňuje vnitřní mikroprostředí kostní dřeně a zhoršuje tak podmínky pro růst nádorových buněk, antiangiogenní aktivita a analgetický účinek.
-
– In vitro: Inhibice osteoblastické proliferace, přímý cytostatický a pro-apoptotický účinek na nádorové buňky, synergický cytostatický účinek spolu s ostatními protinádorovými léky, antiadhezivní/antiinvazivní působení.
Výsledky klinických studií prevence kostních příhod u pacientů s pokročilou formou nádorového onemocnění postihující kosti
První randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie srovnávala kyselinu zoledronovou 4 mg s placebem v prevenci kostních příhod (SREs = Skeletal Related Events) u pacientů s karcinomem prostaty. Kyselina zoledronová 4 mg významně snížila počet pacientů, u kterých došlo k výskytu alespoň jedné kostní příhody (SRE), prodloužila medián času do první SRE o > 5 měsíců a snížila roční výskyt příhod na pacienta – míru onemocnění kostí. Analýzy mnohočetných příhod ukázaly, ve srovnání s placebem, 36 % snížení rizika vývoje SREs ve skupině s kyselinou zoledronovou 4 mg. Pacienti dostávající kyselinu zoledronovou 4 mg hlásili nižší nárůst bolestivosti než ti, kteří dostávali placebo, rozdíl dosáhl významnosti v měsících 3, 9, 21 a 24. U pacientů dostávajících kyselinu zoledronovou 4 mg byl nižší výskyt patologických zlomenin. U pacientů s blastickými lézemi byl léčebný efekt méně zřejmý. Výsledky účinnosti jsou uvedeny v tabulce 2.
V druhé studii, kde byly zahrnuty solidní nádory jiné než karcinomy prsu a prostaty, snížila kyselina zoledronová 4 mg významně počet pacientů s SRE a prodloužila medián času do první kostní příhody na > 2 měsíce a snížila poměr kostní morbidity. Analýza mnohočetných příhod ukázala 30,7 % snížení rizika ve vývoji kostních příhod ve skupině léčené kyselinou zoledronovou 4 mg ve srovnání s placebem. Výsledky účinnosti jsou uvedeny v tabulce 3.
Tabulka 2: Výsledky účinnosti (pacienti s karcinomem prostaty a hormonální léčbou)
| Jakákoliv SRE(+TIH) | Zlomeniny* | Radiační léčba kostí | ||||
| kyselina zoledronová 4 mg | Placebo | kyselina zoledronová 4 mg | Placebo | kyselina zoledronová 4 mg | Placebo | |
| N | 214 | 208 | 214 | 208 | 214 | 208 |
| Procento pacientů s SREs (%) | 38 | 49 | 17 | 25 | 26 | 33 |
| p-hodnota | 0,028 | 0,052 | 0,119 | |||
| Median SRE (dny) | 488 | 321 | NR | NR | NR | 640 |
| p-hodnota | 0,009 | 0,020 | 0,055 | |||
| Míra postižení kostí | 0,77 | 1,47 | 0,20 | 0,45 | 0,42 | 0,89 |
| p-hodnota | 0,005 | 0,023 | 0,060 | |||
| Snížení rizika mnohočetných příhod** (%) | 36 | – | NA | NA | NA | NA |
| p-hodnota | 0,002 | NA | NA | |||
* Zahrnuje vertebrální i nevertebrální zlomeniny
** Všechny kostní příhody, celkový počet stejně jako doba do každé události během studie
NR Nebylo dosaženo
NA Není aplikovatelné
Tabulka 3: Výsledky účinnosti (solidní nádory jiné než nádory prsu a prostaty)
| Jakékoliv SRE (+TIH) | Zlomeniny* | Radiační léčba kostí | ||||
| kyselina zoledronová 4 mg | Placebo | kyselina zoledronová 4 mg | Placebo | kyselina zoledronová 4 mg | Placebo | |
| N | 257 | 250 | 257 | 250 | 257 | 250 |
| Procento pacientů s SREs (%) | 39 | 48 | 16 | 22 | 29 | 34 |
| p-hodnota | 0,039 | 0,064 | 0,173 | |||
| Median SRE (dny) | 236 | 155 | NR | NR | 424 | 307 |
| p-hodnota | 0,009 | 0,020 | 0,079 | |||
| Míra postižení kostí | 1,74 | 2,71 | 0,39 | 0,63 | 1,24 | 1,89 |
| p-hodnota | 0,012 | 0,066 | 0,099 | |||
| Snížení rizika mnohočetných příhod ** (%) | 30,7 | – | NA | NA | NA | NA |
| p-hodnota | 0,003 | NA | NA | |||
* Zahrnuje vertebrální i nevertebrální zlomeniny
** Všechny kostní příhody, celkový počet stejně jako doba do každé události během studie
NR Nebylo dosaženo
NA Nebylo aplikovatelné
V randomizované dvojitě zaslepené studii fáze III, byla srovnávána kyselina zoledronová 4 mg nebo 90 mg pamidronátu při podávání každý 3. až 4. týden pacientům s mnohočetným myelomem nebo karcinomem prsu s nejméně jednou kostní lézí. Výsledky ukázaly, že kyselina zoledronová 4 mg měla v prevenci SREs srovnatelnou účinnost jako 90 mg pamidronátu. Analýza mnohočetných příhod odhalila významné snížení rizika u pacientů léčených kyselinou zoledronovou 4 mg o 16 % ve srovnání s pacienty, kteří dostávali pamidronát. Výsledky účinnosti jsou uvedeny v tabulce 4.
Tabulka 4: Výsledky účinnosti (pacienti s nádorem prsu a mnohočetným myelomem)
| Jakákoliv SRE (+TIH) | Zlomeniny* | Radiační léčba kostí | ||||
| kyselina zoledronová 4 mg | Pam 90 mg | kyselina zoledronová 4 mg | Pam 90 mg | kyselina zoledronová 4 mg | Pam 90 mg | |
| N | 561 | 555 | 561 | 555 | 561 | 555 |
| Procento pacientů s SREs (%) | 48 | 52 | 37 | 39 | 19 | 24 |
| p-hodnota | 0,198 | 0,653 | 0,037 | |||
| Median SRE (dny) | 376 | 356 | NR | 714 | NR | NR |
| p-hodnota | 0,151 | 0,672 | 0,026 | |||
| Míra postižení kostí | 1,04 | 1,39 | 0,53 | 0,60 | 0,47 | 0,71 |
| p-hodnota | 0,084 | 0,614 | 0,015 | |||
| Snížení rizika mnohočetných příhod** (%) | 16 | – | NA | NA | NA | NA |
| p-hodnota | 0,030 | NA | NA | |||
* Zahrnuje vertebrální i nevertebrální zlomeniny
** Všechny kostní příhody, celkový počet stejně jako doba do každé události během studie
NR Nebylo dosaženo
NA Nebylo aplikovatelné
Ve dvojitě zaslepené, randomizované, placebem kontrolované studii byla kyselina zoledronová 4 mg studována u 228 pacientů s dokumentovanými kostními metastázami u karcinomu prsu, byl hodnocen účinek 4 mg kyseliny zoledronové na riziko vzniku kostních příhod (SRE) počítané jako poměr celkového počtu kostních příhod (vyjma hyperkalcemie a po zohlednění předchozích zlomenin) vůči celkovému sledovanému období. Pacienti dostávali po dobu jednoho roku každé čtyři týdny buď 4 mg kyseliny zoledronové nebo placebo. Pacienti byli rovnoměrně rozděleni do skupin léčených kyselinou zoledronovou nebo placebem.
Podíl SRE (kostní příhody/osoba rok) byl 0,628 u kyseliny zoledronové a 1,096 u placeba. Poměr pacientů s nejméně jednou příhodou SRE (vyjma hyperkalcemie) byl 29,8 % ve skupině léčené kyselinou zoledronovou proti 49,6 % ve skupině s placebem (p=0,003). Medián času do zjištění první příhody SRE nebyl v rameni léčby kyselinou zoledronovou na konci studie dosažen a byl signifikantně prodloužen ve srovnání s placebem (p=0,007). Kyselina zoledronová 4 mg snížila riziko příhod SRE o 41 % dle analýz mnohočetných příhod (podíl rizika=0,59, p=0,019) ve srovnání s placebem.
Za 4 týdny léčby ve skupině léčené kyselinou zoledronovou bylo v každém následujícím měření v průběhu léčby pozorováno statisticky významné zlepšení v hodnocení bolesti (použitím Brief Pain Inventory BPI) ve srovnání s placebem (obrázek 1). Hodnocení bolesti ve skupině s kyselinou zoledronovou bylo konzistentně pod úrovní počátečního stavu a bylo provázeno trendem snížení užívání analgetik.
Obrázek 1: Průměrná změna oproti počátečnímu stavu (hodnocena dle BPI). Statisticky významné rozdíly jsou vyznačeny (p<0,05) pro srovnání mezi léčbami (4 mg kyselina zoledronová vs. placebo)
BPI průměrná změna oproti počátečnímu stavu
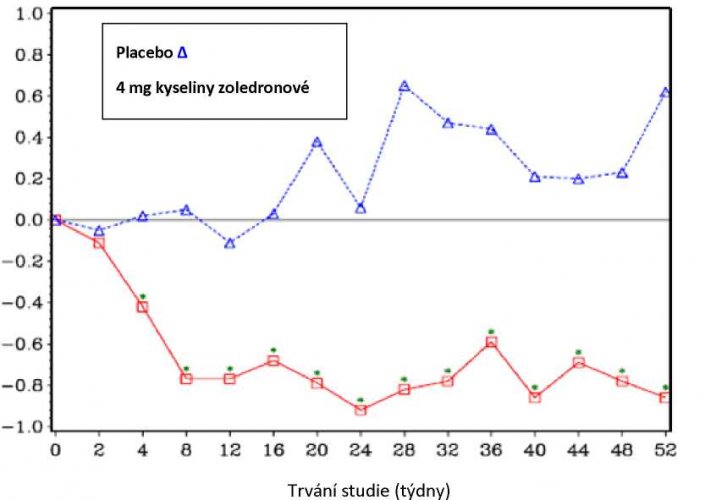
Výsledky klinického hodnocení léčby TIH
V klinických studiích bylo u hyperkalcemie vyvolané nádorovým onemocněním (TIH) demonstrováno, že působení kyseliny zoledronové je charakterizováno poklesem hladiny kalcia v séru a vylučování kalcia močí. Ve studii fáze I pro vyhledávání dávky u pacientů s mírnou až střední hyperkalcemií indukovanou nádorovým onemocněním (TIH), byly testované účinné dávky v rozmezí přibližně 1,22,5 mg.
Aby byly zhodnoceny účinky 4 mg kyseliny zoledronové proti 90 mg pamidronátu, byly výsledky dvou pilotních multicentrických studií u pacientů s TIH kombinovány v předem plánované analýze. Po dávce kyseliny zoledronové 8 mg byla rychlejší normalizace hodnot kalcia v séru pozorována již 4. den a po dávce kyseliny zoledronové 4 mg a 8 mg i 7. den. Byla pozorována následující míra odpovědí:
Tabulka 5: Procento kompletních odpovědí ve dnech v kombinovaných TIH studiích
| Den 4 | Den 7 | Den 10 | |
| Kyselina zoledronová 4 mg (N=86) | 45,3 % (p=0,104) | 82,6 % (p=0,005) | 88,4 % (p=0,002) |
| Kyselina zoledronová 8 mg (N=90) | 55,6 % (p=0,021) | 83,3 % (p=0,010) | 86,7 % (p=0,015) |
| Pamidronát 90 mg (N=99) | 33,3 % | 63,6 % | 69,7 % |
| *p-hodnota značí statistickou významnost proti pamic | ronátu. | ||
Střední doba k dosažení normokalcemie byla 4 dny. Střední doba do relapsu (znovuzvýšení albumin korigované hladiny kalcia v séru na > 2,9 mmol/l) byla u pacientů léčených kyselinou zoledronovou 30 až 40 dnů proti 17 dnům u pacientů léčených pamidronátem 90 mg (p-hodnoty: 0,001 pro 4 mg a 0,007 pro 8 mg kyseliny zoledronové). Mezi dvěma dávkami kyseliny zoledronové nebyl nalezen statistický rozdíl.
-
V klinických studiích 69 pacientů s relapsem nebo refrakterních na počáteční léčbu (kyselina zoledronová 4 mg, 8 mg nebo pamidronát 90 mg) bylo přeléčeno 8 mg kyseliny zoledronové. Frekvence odpovědí byla u těchto pacientů přibližně 52 %. Pro srovnání s dávkou 4 mg kyseliny zoledronové nejsou dostupné údaje, protože tito pacienti byli přeléčeni pouze dávkou 8 mg kyseliny zoledronové.
-
V klinických studiích, provedených u pacientů s hyperkalcemií indukovanou nádorovým onemocněním (TIH), byl celkový bezpečnostní profil mezi všemi 3 léčenými skupinami (kyselina zoledronová 4 a 8 mg a pamidronát 90 mg) podobný v typech i závažnosti.
Pediatrická populace
Výsledky klinické studie v léčbě těžké _ formy osteogenesis imperfecta u pediatrických pacientů ve věku 1 až 17 let
Účinky intravenózně podávané kyseliny zoledronové v léčbě těžké formy osteogenesis imperfecta (typ I, III a IV) u pediatrických pacientů (věk 1 až 17 let) byly srovnány s intravenózně podávaným pamidronátem v jedné mezinárodní, multicentrické, randomizované, otevřené studii se 74 pacienty v rameni s kyselinou zoledronovou a 76 pacienty v rameni s pamidronátem. Doba léčby ve studii byla 12 měsíců, léčbě předcházelo 4–9 týdnů období screeningu, během kterých byla po dobu nejméně 2 týdnů podávána suplementace vitaminu D a kalcia. V klinické studii byli pacienti ve věku 1 až méně než 3 roky léčeni dávkou 0,025 mg/kg kyseliny zoledronové (až do maximální jednotlivé dávky 0,35 mg) každé 3 měsíce a pacienti ve věku 3–17 let byli léčeni dávkou 0,05 mg/kg kyseliny zoledronové (až do maximální jednotlivé dávky 0,83 mg) každé 3 měsíce. Studie byla prodloužena za účelem vyhodnocení dlouhodobé celkové a renální bezpečnosti podávání kyseliny zoledronové jednou nebo dvakrát ročně v průběhu 12 měsíců prodloužené léčby u dětí, které dokončily 1 rok léčby kyselinou zoledronovou nebo pamidronátem v základní studii.
Primárním cílovým parametrem účinnosti ve studii bylo sledování procentuální změny původního stavu kostní denzity bederní páteře (BMD) po 12 měsících léčby. Očekávané účinky léčby na BMD byly podobné, ale design studie nebyl dostatečně robustní k průkazu non-inferiority kyseliny zoledronové. Především nedošlo k jasnému prokázání účinnosti na výskyt zlomenin nebo bolesti. Nežádoucí účinky zlomenin dlouhých kostí dolních končetin byly hlášené u přibližně 24 % (femur) a 14 % (tibia) pacientů s těžkou formou osteogenesis imperfecta léčených kyselinou zoledronovou oproti 12 % a 5 % pacientů s těžkou formou osteogenesis imperfecta léčených pamidronátem, bez ohledu na typ poruchy a kauzalitu, ale souhrnný výskyt zlomenin u pacientů léčených kyselinou zoledronovou a pamidronátem byl srovnatelný: 43 % (32/74) oproti 41 % (31/76). Vyhodnocení rizika zlomenin je ztíženo tím, že zlomeniny jsou častým jevem u pacientů s těžkou formou osteogenesis imperfecta jako součást průběhu onemocnění.
Nežádoucí účinky sledované u této populace byly podobné účinkům dříve zjištěným u dospělých pacientů s pokročilými malignitami ovlivňujícími kosti (viz bod 4.8). Nežádoucí účinky řazené podle četností jsou uvedené v Tabulce 6. Je použita následující konvenční klasifikace: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
Tabulka 6: Nežádoucí účinky sledované u pediatrických pacientů s těžkou formou osteogenesis imperfecta1
| Poruchy nervového systému Časté: | Bolest hlavy |
| Srdeční poruchy Časté: | Tachykardie |
| Respirační, hrudní a mediastinálníporuchy Časté: | Nazofaryngitida |
| Gastrointestinální poruchy Velmi časté: | Zvracení, nauzea |
| Časté: | Bolest břicha |
| Poruchy svalové a kosterní soustavy a pojivové tkáně | |
| Časté: | Bolestivost končetin, artralgie, bolesti svalů a kostí |
| Celkové poruchy a reakce v místě aplikace Velmi časté: | Pyrexie, únava |
| Časté: | Reakce akutní fáze, bolest |
| Vyšetření Velmi časté: | Hypokalcemie |
| Časté: | Hypofosfatemie |
1 Nežádoucí účinky s četností < 5 % byly zhodnoceny z medicínského hlediska a bylo prokázáno, že tyto případy odpovídají stanovenému bezpečnostnímu profilu kyseliny zoledronové, podávané v souladu s indikacemi v bodech 4.1 a 4.2 (viz bod 4.8).
U pediatrických pacientů s těžkou formou osteogenesis imperfecta se zdá být použití kyseliny zoledronové v porovnání s pamidronátem spojené s výraznějším rizikem reakce akutní fáze, hypokalcemie a neobjasněné tachykardie, ale tento rozdíl se snižuje po následných infuzích.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s kyselinou zoledronovou u všech podskupin pediatrické populace při léčbě hyperkalcemie vyvolané nádorovým onemocněním a prevenci kostních příhod u pacientů s pokročilou formou nádorového onemocnění postihujícího kosti (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Následující farmakokinetické údaje získané po jednorázové a opakované 5 a 15 minutové infuzi 2, 4, 8 a 16 mg kyseliny zoledronové u 64 pacientů s kostními metastázami neprokázaly závislost na dávce.
Po zahájení infuze kyseliny zoledronové stoupá rychle koncentrace kyseliny zoledronové v plazmě a dosahuje vrcholu před ukončením infuze, následuje rychlý pokles a za 4 hodiny je koncentrace v plazmě < 10 % vrcholové koncentrace. Za 24 hodin klesne hladina v plazmě < 1 % a pak následuje dlouhé období nízké koncentrace, která nepřesahuje 0,1 % vrcholové koncentrace, před druhou infuzí kyseliny zoledronové 28. den.
Kyselina zoledronová podaná intravenózní infuzí je vylučována třífázovým procesem: rychlé dvojfázové mizení z krevního oběhu s poločasem t/a 0,24 a t/p 1,87 hodiny je následováno dlouhou fází vylučování s terminálním poločasem vylučování t/Y146 hodin. Po opakovaném podání za 28 dnů nebyla pozorována akumulace kyseliny zoledronové v plazmě. Kyselina zoledronová není metabolizována a je vylučována nezměněná ledvinami. Během prvních 24 hodin bylo močí vyloučeno 39 ± 16 % aplikované dávky, zatímco zbytek byl vázán převážně v kostní tkáni. Z kostní tkáně je látka velmi pomalu uvolňována zpět do krevního oběhu a vylučována ledvinami. Celková tělesná clearance je 5,04 ± 2,5 l/h, není závislá na dávce ani ovlivněna pohlavím, věkem, rasou a tělesnou hmotností. Prodloužení doby infuze z 5 na 15 minut způsobí na konci infuze 30% pokles koncentrace kyseliny zoledronové, ale plocha pod křivkou plazmatické koncentrace versus čas není ovlivněna. Doba infuze 20 minut umožňuje dosažení přijatelných maximálních plazmatických koncentrací, aniž by bylo zvýšeno riziko renální toxicity.
Variabilita farmakokinetických parametrů kyseliny zoledronové mezi jednotlivými pacienty je vysoká, stejně jako je tomu u ostatních bisfosfonátů.
U pacientů s hyperkalcemií nebo s jaterní nedostatečností nejsou dostupné farmakokinetické údaje. Kyselina zoledronová neinhibuje lidské P450 enzymy in vitro, nevykazuje biotransformaci a ve studiích na zvířatech bylo < 3 % z aplikované dávky nalezeno ve stolici, což naznačuje, že játra nebudou hrát významnou úlohu ve farmakokinetice kyseliny zoledronové.
Ledvinná clearance kyseliny zoledronové korelovala s clearance kreatininu, ledvinná clearance reprezentuje 75 ± 33 % clearance kreatininu, jejíž průměr byl u 64 studovaných pacientů s nádorovým onemocněním 84 ± 29 ml/min (rozmezí 22 až 143 ml/min). Populační analýza ukázala, že u pacientů s clearance kreatininu 20 ml/min (závažné poškození ledvin), nebo 50 ml/min (středně závažné poškození) je možné odhadnout, že clearance kyseliny zoledronové by byla 37 %, nebo 72 % u pacientů, u nichž byla clearance kreatininu 84 ml/min. U pacientů se závažným postižením ledvin (clearance kreatininu < 30 ml/min) je dostupné jen omezené množství údajů.
V in vitro studii vykazovala kyselina zoledronová nízkou afinitu k buňkám lidské krve s průměrem poměru koncentrace v krvi ke koncentraci v plazmě 0,59 v rozmezí koncentrací 30 ng/ml až 5000 ng/ml. Vazba k plazmatickým proteinům je nízká, s nenavázanou frakcí v rozmezí od 60 % při 2 ng/ml do 77 % při 2000 ng/ml kyseliny zoledronové.
Zvláštní populace
Pediatričtí pacienti
Omezené farmakokinetické údaje u dětí s těžkou formou osteogenesis imperfecta naznačují, že farmakokinetika kyseliny zoledronové je u dětí ve věku 3 až 17 let při obdobném dávkování v mg/kg podobná farmakokinetice u dospělých pacientů. Věk, tělesná hmotnost, pohlaví a clearance kreatininu neměly vliv na systémovou expozici kyseliny zoledronové.
5.3 Předklinické údaje vztahující se k bezpečnosti
Akutní toxicita
Nejvyšší jednorázová intravenózní dávka, kdy nedošlo k žádnému úmrtí zvířat, byla pro myši 10 mg/kg a u potkanů 0,6 mg/kg tělesné hmotnosti.
Subchronická a chronická toxicita
Kyselina zoledronová byla dobře snášena, pokud byla aplikována subkutánně potkanům a intravenózně psům v dávce až 0,02 mg/kg denně po dobu 4 týdnů. Dávka 0,001 mg/kg/den aplikovaná subkutánně potkanům a intravenózní dávka 0,005 mg/kg aplikovaná jednou za 2–3 dny psům po dobu 52 týdnů byla také dobře snášena.
Nejčastějšími nálezy po opakovaném podání studovaných dávek zvířatům v době růstu bylo, téměř po všech dávkách, zvýšení spongiózy v metafýzách dlouhých kostí. Tyto nálezy reflektují farmakologické antiresorpční vlastnosti sloučeniny.
Hranice bezpečnosti z hlediska účinku na ledviny byly při dlouhodobém opakovaném parenterálním podávání experimentálním zvířatům velmi úzké. Kumulativní hladiny bez nežádoucího účinku (NOAELs = no adverse event levels) po jednorázovém podání (1,6 mg/kg) a po opakovaném podávání až po dobu až 1 měsíce (0,06 až 0,6 mg/kg/den) nenaznačily působení na ledviny v dávkách ekvivalentních nebo přesahujících nejvyšší dávky určené pro humánní aplikaci. Dlouhodobé opakované podávání kyseliny zoledronové v dávkách přesahujících nejvyšší předpokládané terapeutické dávky mělo toxické účinky na jiné orgány včetně zažívacího traktu, jater, sleziny, plic a místa intravenózní injekce.
Reprodukční toxicita
Kyselina zoledronová byla při subkutánním podání dávek > 0,2 mg/kg potkanům teratogenní. Ačkoli teratogenita nebo fetotoxicita nebyla pozorována u králíků, byla zjištěna toxicita u březích samic. U potkanů byla pozorována po nejnižší dávce (0,01 mg/kg tělesné hmotnosti) dystokie.
Mutagenita a kancerogenní účinek
V provedených testech mutagenity a kancerogenity nebyl pro kyselinu zoledronovou pozorován ani mutagenní ani kancerogenní účinek.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mannitol
Dihydrát natrium-citrátu
Voda na injekci
6.2 Inkompatibility
Aby se zabránilo možným inkompatibilitám, musí být přípravek Zoledronic acid Fresenius Kabi, koncentrát naředěn 0,9% roztokem chloridu sodného nebo 5% roztokem glukózy.
Tento léčivý přípravek nesmí být mísen s infuzními roztoky obsahujícími kalcium nebo jiné dvojmocné kationty, jako je laktátový Ringerův roztok, a měl by být podáván jako samostatný intravenózní roztok oddělenou infuzní linkou.
6.3 Doba použitelnosti
3 roky
Po naředění: Chemická a fyzikální stabilita po naředění byla prokázána po dobu 24 hodin při teplotě 2°C-8°C. Z mikrobiologického hlediska má být naředěný infuzní roztok použit okamžitě. Pokud není použit okamžitě, je doba uchovávání a podmínky před použitím v plné zodpovědnosti uživatele. Doba uchovávání by neměla být delší než 24 hodiny při teplotě 2°C – 8°C. Chlazený roztok musí být před podáním temperován na pokojovou teplotu.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání naředěného léčivého přípravku viz bod 6.3.
6.5 Druh obalu a velikost balení
Plastová injekční lahvička z bezbarvého polypropylenu uzavřená bromobutylovou pryžovou zátkou a hliníkovým víčkem s plastovým flip-off uzávěrem.
Balení obsahuje 1, 4 nebo 10 injekčních lahviček.
Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Před aplikací musí být 5,0 ml koncentrátu z jedné injekční lahvičky nebo obsah koncentrátu odebraný dle potřeby dále naředěn ve 100 ml roztoku, který neobsahuje kalcium (0,9% roztok chloridu sodného nebo 5% roztok glukózy).
Studie se skleněnými lahvičkami, stejně jako s několika nádobami z polyvinylchloridu, polyethylenu a polypropylenu (předplněnými 0,9% roztokem chloridu sodného nebo 5% roztokem glukózy) neprokázaly inkompatibilitu s kyselinou zoledronovou.
Další informace o nakládání s přípravkem Zoledronic acid Fresenius Kabi zahrnující instrukce o přípravě redukovaných dávek jsou uvedeny v bodě 4.2.
Během přípravy infuze musí být dodržen aseptický postup. Pouze pro jednorázové použití.
Smí být použit pouze čirý, bezbarvý roztok bez částic.
Zdravotnickým pracovníkům se důrazně doporučuje, aby nelikvidovali nevyužitý přípravek Zoledronic acid Fresenius Kabi v lokálním odpadovém systému.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Fresenius Kabi s.r.o.
Na Strži 1702/65, Nusle, 140 00 Praha 4, Česká republika
8. REGISTRAČNÍ ČÍSLO
87/056/13-C
Další informace o léčivu ZOLEDRONIC ACID FRESENIUS KABI
Jak
se ZOLEDRONIC ACID FRESENIUS KABI
podává: intravenózní podání - koncentrát pro infuzní roztok
Výdej
léku: na lékařský předpis
Balení: Injekční lahvička
Velikost
balení: 1X5ML
Držitel rozhodnutí o registraci daného léku v České republice:
Fresenius Kabi s.r.o., Praha
E-mail: marina.svobodova@fresenius-kabi.com
Telefon: +420 225 270 583