Souhrnné informace o léku - CERNEVIT
1. NÁZEV PŘÍPRAVKU
CERNEVIT prášek pro injekční nebo infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
| 1 lahvička (5 ml) obsahuje: | |
| Retinoli palmitas | 3 500 IU |
| (odp. Vitamín A | 1,925 mg) |
| Colecalciferolum | 220 IU |
| (odp. Vitamín D3 | 0,0055 mg) |
| Tocoferolum alfa RRR | 10,200 mg |
| (odp. Vitamín E | 11,200 IU) |
| Acidum ascorbicum (odp. Vitamín C) | 125,000 mg |
| Cocarboxylasum tetrahydricum | 5,800 mg |
| (odp. Vitamín B1 | 3,510 mg) |
| Riboflavini natrii phosphas dihydricus | 5,670 mg |
| (odp. Vitamín B2 | 4,140 mg) |
| Pyridoxini hydrochloridum | 5,500 mg |
| (odp. Vitamín B6 | 4,530 mg |
| Cyanocobalaminum (odp. Vitamín B12) | 0,006 mg |
| Acidum folicum (odp. Kyselina listová) | 0,414 mg |
| Dexpanthenolum | 16,150 mg |
| (odp. Kyselina pantothenová | 17,250 mg) |
| Biotinum (odp. Biotin) | 0,069 mg |
| Nicotinamidum (odp. Vitamín PP) | 46,000 mg |
Pomocná látka se známým účinkem: sójové fosfatidy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro injekční nebo infuzní roztok.
Popis přípravku: oranžovo-žlutý lyofilizát, bez zápachu až téměř bez zápachu.
4. KLINICKÉ ÚDAJE
4.1. Terapeutické indikace
Doplnění vitamínů u pacientů na parenterální výživě.
Pouze pro dospělé a děti starší 11 let.
4.2. Dávkování a způsob podání
Dospělí, dospívající a děti starší 11 let
1 lahvička denně
4.3. Kontraindikace
Přípravek CERNEVIT nesmí být podáván:
u hypersenzitivity na léčivé látky, zejména na vitamín B1, nebo na kteroukoli pomocnou látku (uvedenou v bodě 6.1), zahrnující sójové proteiny / přípravky (lecithin ve smíšených micelách je sójového původu) nebo arašídové proteiny / přípravky novorozencům, kojencům a dětem mladším 11 let u hypervitaminózy (zejména vitamínu A, E a D3)-
4.4. Zvláštní upozornění a zvláštní opatření pro použití
Hypersenzitivní reakce
- U přípravku CERNEVIT byly hlášeny mírné až závažné systémové hypersenzitivní reakce na složky přípravku (včetně vitamínů B1, B2, B12, kyselinu listovou a sójový lecithin) (viz bod 4.8).
- V některých případech mohou být projevy hypersenzitivní reakce během intravenózního podávání multivitamínů závislé na rychlosti podávání. Při intravenózní infuzi je zapotřebí přípravek CERNEVIT podávat pomalu. Při intravenózní injekci je třeba injekci aplikovat pomalu (v průběhu alespoň 10 minut).
- Infuzi nebo injekci je zapotřebí okamžitě ukončit, pokud se vyskytnou projevy či příznaky hypersenzitivní reakce.
- Aby nedošlo k předávkování a toxickým účinkům, je zapotřebí monitorovat klinický stav pacienta a koncentrace vitamínů v krvi, obzvláště u vitamínů A, D a E, a to především u pacientů, kteří přijímají navíc vitamíny z jiných zdrojů, nebo užívají jiné přípravky zvyšující riziko vitamínové toxicity.
- Monitorování je důležité především u pacientů podstupujících dlouhodobou suplementaci.
Hypervitaminóza A
– Riziko hypervitaminózy A a toxicita vitamínu A (např. abnormality kůže a kostí,
diplopie, cirhóza) je vyšší například u pacientů s proteinovou malnutricí, poškozením ledvin (i při nepřítomnosti suplementace vitamínem A), poškozením jater, pacientů s drobnou tělesnou konstitucí (např. u pediatrických pacientů) a pacientů na chronické terapii.
– Akutní jaterní onemocnění u pacientů se saturovanými zásobami vitamínu A
může vést k manifestaci toxicity vitamínem A.
Hypervitaminóza D
– Nadměrné množství vitamínu D může způsobit hyperkalcémii a hyperkalcinurii
– Riziko toxicity vitamínem D je zvýšeno u pacientů s hyperparathyroidismem nebo u pacientů s dlouhodobým podáváním vitamínů.
Hypervitaminóza E
– I když velmi vzácně, nadměrné dávky vitamínu E nepříznivě ovlivňují hojení rány a podílejí na dysfunkci krevních destiček
– Riziko toxicity vitamínem E je zvýšeno u pacientů s poruchou jater, u pacientů, kteří mají poruchu krvácivosti nebo jsou na perorálních antikoagulanciích nebo u pacientů s dlouhodobým podáváním vitamínů.
„Refeeding“ syndrom u pacientů, kterým je podávána parenterální výživa
Realimentace velmi nedostatečně živených pacientů může způsobit “refeeding“ syndrom, pro který je charakteristický nitrobuněčný posun draslíku, fosforu a hořčíku, zatímco se pacient stává anabolickým. Může se také rozvinout deficit thiaminu a zadržování tekutin. Pečlivé sledování a pomalé zvyšování příjmu živin při současné eliminaci nadměrného podávání živin může vzniku těchto komplikací zabránit. Při deficitu živin může být opodstatněná odpovídající suplementace.
Precipitáty u pacientů, kterým je podávána parenterální výživa
Plicní vaskulární precipitáty byly hlášeny u pacientů přijímajících parenterální výživu. V některých případech došlo k fatálním následkům. Nadměrné přidávání vápníku a fosfátů zvyšuje riziko tvorby kalcium-fosfátového precipitátu. Precipitáty byly hlášeny dokonce, i když fosfátová sůl nebyla v roztoku přítomna. Byly hlášeny také sraženiny distálně od in-line filtru a suspektní precipitáty v krevním řečišti.
Kromě kontroly roztoku je také třeba pravidelně kontrolovat, zda se sraženiny nenacházejí v infuzní soupravě a v katétru.
Pokud se objeví příznaky plicních potíží, je zapotřebí infuzi zastavit a zahájit lékařské zhodnocení.
Zkontrolujte, zda obal není porušen.
Dodržujte zásady aseptické techniky.
Nepoužívejte částečně použité lahvičky nebo přípravek, který je po rozpuštění neobvykle zabarven.
Účinky na játra
- U pacientů užívajících přípravek CERNEVIT se doporučuje monitorovat parametry jaterní funkce. Obzvláště pečlivé monitorování se doporučuje u pacientů se žloutenkou nebo jiným důkazem cholestázy.
- Je známé, že u některých pacientů na parenterální výživě (včetně parenterální výživy doplněné vitamíny) dochází k rozvoji hepatobiliárních poruch včetně cholestázy, steatózy jater, fibrózy a cirhózy, které mohou vést k jaternímu selhání, stejně jako k cholecystitidě a cholelitiáze. Etiologie těchto poruch se považuje za multifaktoriální a u jednotlivých pacientů se může lišit. Pacienty s rozvojem abnormálních laboratorních parametrů nebo jiných příznaků hepatobiliárních poruch by měl včas zhodnotit lékař -hepatolog, který může identifikovat případné kauzální a kontribuční faktory a možnou terapeutickou a profylaktickou intervenci.
Použití u pacientů s poškozenou funkcí jater
Pacienti s poškozením jater mohou vyžadovat individualizovanou vitamínovou suplementaci. Zvláštní pozornost je třeba věnovat prevenci toxicity vitamínem A, protože aktuální jaterní onemocnění je spojeno se zvýšeným sklonem k toxicitě vitamínem A, především v kombinaci s chronicky nadměrnou spotřebou alkoholu (viz také části „Hypervitaminóza A“ a „Účinky na játra“ výše).
Použití u pacientů s poškozením funkce ledvin
Pacienti s poškozením ledvin mohou vyžadovat individualizovanou vitamínovou suplementaci, v závislosti na míře poškození ledvin a průvodním zdravotním stavu. U pacientů se závažným poškozením funkce ledvin je třeba věnovat zvláštní pozornost udržování odpovídajícího stavu vitamínu D a zabránit toxicitě vitamínu A, která se může projevit u pacientů suplementovaných nízkou dávkou vitamínu A, nebo i bez suplementace. Hypervitaminóza a toxicita pyridoxinu (vitamín B6) (periferní neuropatie, mimovolní pohyby) byla hlášena u pacientů na chronické hemodialýze, kterým jsou podávány nitrožilní multivitamíny obsahující 4 mg pyridoxinu podávaného třikrát týdně.
Všeobecný monitoring
U pacientů, kterým jsou po delší dobu podávány parenterální multivitamíny jako jediný zdroj vitamínů, je zapotřebí sledovat klinický stav a hladiny vitamínů. Zejména důležité je monitorovat odpovídající suplementaci například:
- vitamínu A u pacientů s proleženinami, poraněními, spáleninami, syndromem krátkého střeva nebo cystickou fibrózou
- vitamínu B1 u pacientů na dialýze
- vitamínu B2 u pacientů s rakovinou
- vitamínu B6 u pacientů s poškozením ledvin
- jednotlivých vitamínů, jejichž potřeba může být zvýšena kvůli interakcím s jinými léky (viz bod 4.5).
Deficit jednoho nebo více vitamínů musí být upraven specifickou suplementací.
Přípravek CERNEVIT neobsahuje vitamín K, který je v případě potřeby nutno podat samostatně.
Použití u pacientů s deficitem vitamínu B12
Před zahájením podávání přípravku CERNEVIT se doporučuje pacientům ohroženým deficitem vitamínu B12 a/nebo pokud je plánována suplementace přípravkem CERNEVIT po několik týdnů, vyhodnotit stav vitamínu B12.
Po několika dnech podávání mohou být jednotlivá množství kyanokobalaminu (vitamín B12) a kyseliny listové v přípravku CERNEVIT dostačující, následkem čehož se u některých pacientů s megaloblastickou anémií spojenou s deficitem vitamínu B12 zvyšuje počet červených krvinek, počtu retikulocytů a hodnot hemoglobinu. To může maskovat aktuální deficit vitamínu B12. Účinná léčba deficitu vitamínu B12 vyžaduje vyšší dávky kyanokobalaminu, než které přípravek CERNEVIT obsahuje.
Suplementace kyseliny listové u pacientů s deficitem vitamínu B12, kteří současně neužívají vitamín B12, nezabraňuje rozvoji ani progresi neurologických projevů spojených s deficitem vitamínu B12. Nabízí se, že se neurologické zhoršení může urychlit.
Při interpretaci hladin vitamínu B12 by se mělo vzít v úvahu, že příjem vitamínu B12 v nedávné době může vést k normálním hladinám navzdory tkáňovému deficitu.
Interference laboratorních testů
V závislosti na použitých reagentech může přítomnost kyseliny askorbové v krvi a moči způsobit falešně vysoké nebo nízké hladiny glukózy v některých testech moči a krve, včetně testovacích proužků a ručních glukometrů. Technické informace všech laboratorních testů je třeba konzultovat, aby bylo možné stanovit potenciální interferenci způsobenou vitamíny.
Současné podávání anti-epileptických léčivých přípravků
Vzhledem k obsahu kyseliny listové v přípravku CERNEVIT je třeba dodržovat zvláštní opatření při kombinaci s léčbou antiepileptiky obsahujícími fenobarbital, fenytoin nebo primidon (viz bod 4.5 Interakce s jinými léčivými přípravky a jiné formy interakce).
Současné podávání s Levodopou
Vzhledem k obsahu pyridoxinu v přípravku CERNEVIT je třeba dodržovat zvláštní opatření při kombinaci s léčbou levodopou, protože může snížit účinek L-dopy (viz bod 4.5 Interakce s jinými léčivými přípravky a jiné formy interakce).
Pediatrická populace
Přípravek CERNEVIT je indikován u pediatrických pacientů starších 11 let (viz také bod 4.4: Hypervitaminóza A výše).
Geriatrická populace
Obecně je vhodné u starších pacientů zvážit upravení dávky (snížení dávky a/nebo prodloužení dávkovacích intervalů) odrážející vyšší frekvenci poklesu funkce jater, ledvin nebo srdce, stejně jako průvodních onemocnění nebo užívané medikace.
Obsah sodíku
Přípravek CERNEVIT obsahuje 24 mg sodíku (1 mmol) v každé lahvičce. To je třeba vzít v úvahu u pacientů na kontrolované nízkosodíkové dietě.
Před smícháním s jinými infuzními roztoky je třeba zkontrolovat kompatibilitu, zvláště je-li přípravek CERNEVIT přidáván do vaků s binární směsí pro parenterální výživu kombinující glukózu, a roztok elektrolytů a aminokyselin, případně do vaků s vícesložkovou směsí kombinující glukózu, roztok elektrolytů a aminokyselin a lipidy (viz také bod 6.2 Inkompatibility).
4.5. Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce mezi specifickými vitamíny v přípravku CERNEVIT a dalšími přípravky je třeba odpovídajícím způsobem řešit.
K těmto interakcím patří:
- Přípravky, které mohou způsobovat pseudotumor cerebri (idiopatická intrakraniální hypertenze) (včetně určitých tetracyklinů): Zvýšené riziko idiopatické intrakraniální hypertenze při současném podávání vitamínu A.
- Alkohol (chronická nadměrná konzumace): Zvyšuje riziko hepatotoxicity vitamínu A
- Antikonvulziva: Kyselina listová může zvyšovat metabolismus některých antiepileptik, např. fenobarbitalu, fenytoinu, fosfenytoinu a primidonu, což může zvyšovat riziko záchvatů. Při současném podávání folátů a po jeho ukončení je třeba pečlivě monitorovat plazmatické koncentrace antikonvulziv.
- Vitamín E může podporovat inhibici funkce krevních destiček nebo zesílit antikoagulační účinek přípravků jako jsou aspirin nebo warfarin.
- Kyselina acetylsalicylová (léčba vysokými dávkami) Může snižovat hladiny kyseliny listové zvýšením exkrece močí.
- Určitá antikolvuziva (např. fenytoin, karbamazepin, fenobarbital, valproát): Mohou způsobovat deficit folátu, pyridoxinu a vitamínu D
- Určitá antivirotika: Snížené hladiny vitamínu D jsou spojovány např. s přípravky efavirenz a zidovudin. Snížená tvorba aktivních metabolitů vitamínu D byla spojena s inhibitory proteázy.
- Chloramfenikol: Může inhibovat hematologickou odpověď na léčbu vitamínem B12.
- Deferoxamin: Zvýšené riziko srdečního selhání způsobeného železem z důvodu zvýšené mobilizace železa při suprafyziologické suplementaci vitamínu C. Specifická opatření viz informace o přípravku deferoxamin.
- Etionamid: Může způsobovat deficit pyridoxinu.
- Fluoropyrimidiny (5-fluorouracil, kapecitabin, tegafur): Zvýšená toxicita při kombinaci s kyselinou listovou.
- Antagonisté folátu, např. metotrexát, sulfasalazin, pyrimetamin, triamteren, trimetoprim a vysoké dávky čajových katechinů: blokování konverze folátů na jejich aktivní metabolity a snížení účinnosti suplementace
- Antimetabolity folátu (metotrexát, raltitrexed): Suplementace kyselinou listovou může mít za následek potřebu vyšší dávky těchto látek, aby se dosáhlo požadovaného terapeutického účinku.
- Levodopa: Vitamín B6 může snižovat účinnost levodopy, protože dekarboxylace levodopy vyžaduje enzym závislý na vitamínu B6. Pro zabránění této interakci lze přidat dopa-dekarbolyzovaný inhibitor, jako je karbidopa.
- Antagonisté pyridoxinu, včetně cykloserinu, hydralazinu, isoniazidu, penicillaminu, fenelzinu: Mohou způsobovat deficit pyridoxinu.
- Retionidy, včetně bexarotenu: Zvyšují riziko toxicity, pokud jsou používány současně s vitamínem A (viz bod 4.4: Hypervitaminóza A)
- Theofylin: Může způsobovat deficit pyridoxinu
Léky navazující se na alfa1-kyselý glykoprotein (AAG):
U pacientů užívajících přípravek CERNEVIT společně s přípravky vážící se na AAG je zapotřebí pečlivě sledovat potenciálně zvýšenou reakci na tyto přípravky, mezi které patří propranolol, prazosin a mnohé další.
Interakce s jinými vitamínovými doplňky:
Některé přípravky mohou interagovat s určitými vitamíny v dávkách podstatně vyšších, než jsou dávky poskytované přípravkem CERNEVIT. To je třeba zvážit u pacientů, kteří dostávají vitamíny z více zdrojů, a v případě potřeby je vhodné u pacientů takové interakce podrobně sledovat a odpovídajícím způsobem řešit.
4.6. Fertilita, těhotenství a kojení
Před předepsáním přípravku CERNEVIT by měli lékaři pečlivě zvážit možná rizika a benefity pro každého konkrétního pacienta.
Údaje o bezpečnosti podávání přípravku CERNEVIT v období těhotenství a kojení nejsou k dispozici. Přípravek může být předepsán během těhotenství, je-li to zapotřebí, za předpokladu, že indikace a dávkování jsou sledovány, aby se předešlo předávkování vitamínů.
Použití během kojení se nedoporučuje vzhledem k možnosti předávkování novorozence vitamínem A.
Nejsou k dispozici adekvátní data o podávání přípravku CERNEVIT v souvislosti s fertilitou mužů a žen.
4.7. Účinky na schopnost řídit a obsluhovat stroje
Neuplatňuje se.
4.8. Nežádoucí účinky
Nežádoucí účinky (ADR), které se vyskytly po podání přípravku CERNEVIT jsou uvedeny s jejich relativní četností; mezi ně patří nežádoucí účinky (ADR) popsané v klinických studiích a z post marketingového hlášení. Přípravek CERNEVIT byl podáván v průběhu 3 klinických studií 267 dospělým pacientům vyžadujících parenterální vitamínový doplněk.
Frekvence nežádoucích účinků jsou hlášeny použitím následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000); a není známo (z dostupných údajů nelze určit).
Nežádoucí účinky z klinických studií a post marketingového sledování hlášené k přípravku
CERNEVIT:
| Třídy orgánových systémů | Upřednostňovaný termín MedDRA | Frekvence |
| Poruchy imunitního systému | Systémové reakce hypersensitivity s projevy jako jsou respirační potíže, nepříjemné pocity na hrudi, stažení hrdla, tachypnoe, kopřivka, | vzácné |
| vyrážka, erytém, nepříjemné pocity v oblasti epigastria a tachykardie Anafylaktická reakce | není známo | |
| Poruchy metabolismu a výživy | Zvýšený vitamín Aa,b Zvýšená hladina proteinu vázajícího retinol | není známoc není známoc |
| Poruchy nervového systému | Poruchy chuti (kovová příchuť) | není známo |
| Gastrointestinální poruchy | Nauzea Zvracení | méně časté méně časté |
| Průjem | není známo | |
| Poruchy jater a žlučových cest | Zvýšené transaminázy Zvýšená glutamát dehydrogenáza Zvýšená alkalická fosfatáza v krvi Zvýšené žlučové kyselinye | není známoc není známoc není známoc není známoc |
| Poruchy kůže a podkožní tkáně | Svědění | není známo |
| Celkové poruchy a reakce v místě aplikace | Bolest v místě vpichu injekce/infuze | časté |
| Horečka Generalizovaná bolest Reakce v místě aplikace infuze, např. pálení, vyrážka | není známo není známo není známo |
aNehlášeny žádné příznaky hypervitaminózy vitamínu A.
bVyšší hladiny vitamínu A v plazmě byly 45. den podávání hlášeny u 8 z 20 pacientů, kterým byl přípravek CERNEVIT podáván v parenterální výživě. Od 45. do 90. dne podávání výrobku zůstávaly vysoké dávky vitamínu A stabilní (maximální pozorovaná hodnota 3,6 pmol/l v 90. den; běžné hodnoty: 1 až 2,6 pmol/l). Kromě toho bylo také zjištěno zvýšení bílkoviny vážící retinol (RBP). Byla hlášena maximální pozorovaná hodnota RBP 60 mg/l v 90. den (normální hodnoty: 30 až 50 mg/l). c Četnost buď nelze stanovit, nebo je celkový počet pacientů v jednotlivých studiích natolik nízký, že neumožňuje platné stanovení četnosti.
d Izolované zvýšení alaninaminotransferázy bylo hlášeno při současně probíhajícím zánětlivém onemocnění střev. Přípravek CERNEVIT byl podán intravenózní injekcí bez parenterální výživy.
e Zvýšení celkových a jednotlivých žlučových kyselin včetně kyseliny glykocholové bylo hlášeno u parenterální výživy po jejím zahájení u pacientů dostávajících přípravek CERNEVIT.
Hypersenzitivní reakce na složky jsou především v důsledku alergických reakcí na vitamín B1. Intenzita se může lišit od velmi mírných až po závažné alergické reakce.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu: Státní ústav pro kontrolu léčiv, Šrobárova 48, 100 41 Praha 10.
Webové stránky:
4.9. Předávkování
Akutní nebo chronické předávkování vitamínů (zejména A, B6, D a E) může způsobit symptomatické hypervitaminózy.
Riziko předávkování je zvlášť vysoké, jestliže pacient dostává vitamíny z více zdrojů a celkové doplnění vitamínů neodpovídá individuálním požadavkům pacienta a u pacientů se zvýšeným sklonem k hypervitaminóze (viz bod 4.4).
Příznaky předávkování jsou nejčastěji způsobeny podáním nadměrných dávek vitamínu A.
Klinické příznaky akutního předávkování vitamínem A (dávky převyšující 150, 000 IU)
- gastrointestinální obtíže, bolesti hlavy, intrakraniální hypertenze, edém papily, psychiatrické příznaky, iritabilita až křeče, zpožděná generalizovaná deskvamace.
Klinické příznaky chronické intoxikace (riziko při dlouhodobém podávání vitamínu A v nadměrných dávkách osobám bez deficitu)
- intrakraniální hypertenze, kortikální hyperostóza dlouhých kostí. Diagnóza je založena zpravidla na přítomnosti bolestivých subkutánních otoků končetin. RTG vyšetření potvrzuje diafyzální periostální ztluštění ulny, fibuly, klavikul a žeber.
Léčba předávkování vitamínem znamená obvykle jeho vysazení a další opatření, která jsou klinicky indikována, jako je snížení příjmu vápníku, zvýšení diurézy a rehydratace.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1, Farmakodynamické vlastnosti
Farmakoterapeutická skupina: vitamíny
ATC kód: B05XC
Přípravek CERNEVIT poskytuje vyvážený obsah vitamínů rozpustných ve vodě a v tucích odpovídající denní potřebě při parenterální výživě.
Farmakodynamické účinky přípravku CERNEVIT se shodují s farmakodynamickými účinky 12 jednotlivých vitamínů obsažených v přípravku.
Základní vlastnosti jsou následující:
| Vitamín A: | ovlivňuje buněčný růst a diferenciaci a fyziologické mechanismy vidění. |
| Vitamín D: | reguluje metabolismus kalcia a fosforu v kostech a ledvinách. |
| Vitamín E: | antioxidační vlastnosti, předchází tvorbě toxických oxidačních produktů a chrání buněčné složky. |
| Vitamín B1 (thiamin): | v kombinaci s ATP je koenzymem zasahujícím do metabolismu cukrů. |
| Vitamín B2 (riboflavin): | působí jako koenzym v buněčném energetickém metabolismu, tkáňových respiračních systémech a metabolismu výživy. |
| Vitamín B3 (PP): | jako složka koenzymů NAD a NADP v oxidačně-redukčních reakcích je nezbytný pro metabolismus výživy a tkáňové dýchání. |
| Vitamín B5 (kys. pantothenová): | prekurzor koenzymu A, spojen s oxidativním metabolismem cukrů, glukoneogenezou, syntézou mastných kyselin, sterolů, steroidních hormonů a porfyrinů. |
| Vitamín B6 (pyridoxin): | působí jako koenzym v metabolismu bílkovin, cukrů a tuků. |
| Vitamín B12: | z exogenních zdrojů, potřebný pro syntézu nukleoproteinu a myelinu, buněčnou reprodukci, normální růst a udržení normální erytropoézy. |
| Vitamín C: | antioxidační vlastnosti, nezbytný pro tvorbu a udržování nitrobuněčné substance a kolagenu, biosyntézu katecholaminů, syntézu karnitinu a steroidů, metabolismus kyseliny listové a tyrosinu. |
| Kyselina listová: | z exogenních zdrojů, potřebná pro syntézu nukleoproteinů a udržení normální erytropoézy. |
| Biotin: | ve vazbě na nejméně čtyři enzymy se účastní energetického metabolismu, včetně glukoneogenezy. |
5.2. Farmakokinetické vlastnosti
U pacientů, kterým je podáván přípravek CERNEVIT, se obnovují plazmatické hladiny vitamínů A, D a E a během dlouhodobé parenterální výživy se tyto hladiny udržují v normálním rozmezí.
Farmakokinetické vlastnosti přípravku CERNEVIT se shodují s farmakokinetickými vlastnostmi 12 individuálních vitamínů obsažených v přípravku. Základní vlastnosti jsou následující:
| Vitamín A: | normální sérová hladina 80–300 IU/ml, váže se na protein, vylučován zejména žlučí a také močí. |
| Vitamín D: | aktivní po hydroxylaci v játrech a ledvinách, váže se na protein, primárně vylučován žlučí a močí. |
| Vitamín E: | přenášený v krvi lipoproteiny, v játrech přeměňován na lakton a vylučován především močí. |
| Vitamín B1 (thiamin): | 90% je přenášeno erytrocyty, v plazmě je většinou vázán na albumin, vylučován hlavně močí. |
| Vitamín B2 (riboflavin): | vázán na plazmatické bílkoviny, kolísavé plazmatické hladiny, vylučován zejména močí ve volné podobě nebo v podobě metabolitů. |
| Vitamín B3 (PP): | v plazmě ve formě kyselin a amidů; vylučován ve volné podobě nebo v podobě metabolitů. |
| Vitamín B5 (kys. pantothenová): | obsažen ve volné formě nebo jako koenzym A v plazmě a erytrocytech, vylučován močí. |
| Vitamín B6 (pyridoxin): | metabolizován v játrech, vylučován močí. |
| Vitamín B12: | normální sérové hladiny 200–900 pg/ml; vázán na proteiny, uchováván v játrech, přechází do mléka, 50–90% dávky je vylučováno močí. |
| Vitamín C: | v běžných koncentracích (8–14 mg/l) dochází k totální renální tubulární reabsorpci, nadlimitní koncentrace jsou vylučovány močí. |
| Kyselina listová: | normální sérové koncentrace v rozmezí 0,005–0,015 pg/ml; distribuována do všech tkání, je metabolizována a uchovávána v játrech, ve vysokých dávkách, je-li překročena renální tubulární reabsorpce, vylučuje se močí. |
| Biotin: | v plazmě se objevuje ve vázané nebo volné podobě, uchováván především v játrech, vylučuje se většinou nezměněn močí. |
5.3. Předklinické údaje vztahující se k bezpečnosti
Nebyly prováděny specifické předklinické studie s přípravkem CERNEVIT.
Předklinické studie bezpečnosti jednotlivých složek přípravku CERNEVIT nesvědčí pro rizika při podávání lidem.
6. FARMACEUTICKÉ ÚDAJE
6.1. Seznam pomocných látek
glycin
kyselina glykocholová
sójové fosfatidy
roztok hydroxidu sodného 1 mol/l nebo roztok kyseliny chlorovodíkové 1 mol/l k úpravě pH
6.2. Inkompatibility
- Aditiva mohou být nekompatibilní s parenterální výživou obsahující přípravek CERNEVIT.
- Před přidáním jiných léčivých přípravků nebo látek nejprve potvrďte jejich kompatibilitu a stabilitu výsledného přípravku.
- Pokud je nutné podávat spojkou Y-site přípravky, které jsou nekompatibilní, podávejte je oddělenými IV linkami.
- Vitamín A a thiamin v přípravku CERNEVIT mohou reagovat s bisulfity v roztocích parenterální výživy (např. kvůli příměsím), což vede k degradaci vitamínu A a thiaminu.
- Zvýšené pH roztoku může zvýšit degradaci některých vitamínů. To je třeba zvážit při přidávání zásaditých roztoků ke směsím obsahujícím přípravek CERNEVIT.
- Stabilita kyseliny listové může být porušena zvýšenou koncentrací vápníku ve směsi.
- Byly popsány mnohé další inkompatibility mezi vitamíny a jinými léčivými přípravky, včetně určitých antibiotik a stopových prvků.
6.3. Doba použitelnosti
2 roky
Po naředění byla prokázána chemická a fyzikální stabilita po dobu 24 hodin při teplotě 25 °C. Z mikrobiologického hlediska má být přípravek použit ihned po rozpuštění. Není-li použit okamžitě, doba a podmínky uchovávání přípravku před použitím jsou v odpovědnosti uživatele a doba by neměla být delší než 24 hodin při teplotě 2 °C – 8 °C, pokud rozpuštění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4. Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Uchovávejte lahvičky v krabičce, aby byl přípravek chráněn před světlem.
6.5. Druh obalu a velikost balení
-
a) lahvička z hnědého skla s pryžovou zátkou s Al krytem, přířez, krabička s obsahem 1,
10 nebo 20 lahviček
-
b) lahvička z hnědého skla s pryžovou zátkou a BIO-SETEM, krabička s obsahem 1, 10 nebo 20 lahviček
Na trhu nemusí být všechny velikosti balení.
6.6. Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
- Během rekonstituce musejí být zachovány aseptické podmínky, pokud je přípravek používán jako součást směsi v parenterální výživě.
- Jemně míchejte, aby se rozpustil lyofilizovaný prášek.
- Pro další použití musí být přípravek CERNEVIT v lahvičce zcela rozpuštěn.
- Přípravek nepoužívejte, pokud rekonstituovaný roztok není čirý a originální těsnění neporušené.
- Po přidání přípravku CERNEVIT do parenterálního roztoku zkontrolujte, zda nedošlo k jakékoli abnormální změně zbarvení a/nebo výskytu precipitátů, nerozpustných komplexů nebo krystalů.
- Pokud se přípravek CERNEVIT používá jako součást směsi v parenterální výživě, důkladně smíchejte výsledný roztok.
- Veškerý nevyužitý podíl rekonstituovaného přípravku CERNEVIT je třeba zlikvidovat a nesmí se uchovávat pro další míchání.
- Před podáním je třeba u parenterálních léčivých přípravků vizuální kontrola na přítomnost jakýchkoli částeček a abnormálního zbarvení, pokud to roztok a obal umožňují.
- Během podávání roztoků parenterální výživy je doporučeno používat konečný filtr.
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou případů, ve kterých byla kompatibilita a stabilita prokázána (viz bod 4.2). V tomto případě prosím kontaktujte pro další informace držitele rozhodnutí o registraci.
Kompatibilita s roztoky podávanými současně stejnou linkou musí být zkontrolována (viz bod 6.2).
Přípravek CERNEVIT (lahvička bez aplikační sady BIO-SET)
Pomocí injekční stříkačky naplňte lahvičku 5 ml vody na injekci nebo 5 % roztoku glukózy nebo 0,9% roztoku chloridu sodného.
Lehce promíchejte, aby došlo k rozpuštění prášku.
Výsledný roztok je oranžovo-žluté barvy.
PŘÍPRAVEK CERNEVIT BIO-SET
Přípravek CERNEVIT BIO-SET umožňuje přímé rozpouštění ve vacích (jednokomorových nebo vícekomorových plastových vacích) vybavených injekčním vstupem.
Jednokomorové vaky:
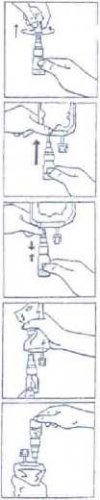
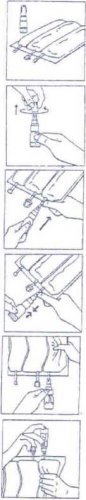
-
1. Sejměte uzávěr pootočením a následným tahem za jisticí pruh uzávěru.
-
2. Aplikační sadu BIO-SET napojte na injekční vstup.
-
3. Tlakem na průhlednou pohyblivou část aplikační sady propíchněte pryžový uzávěr lahvičky.
-
4. Propojený vak a aplikační sadu držte ve vertikální poloze vakem nahoru. Několikrát lehce stlačte vak, čímž naplníte lahvičku (přibližně 5 ml) roztokem. Protřepáním lahvičky dojde k rozpuštění přípravku CERNEVIT.
-
5. Napojený vak a aplikační sadu obraťte vzhůru nohama. Několikrát lehce stlačte vak, čímž dojde k vytlačení vzduchu do lahvičky a roztoku do vaku.
-
6. Body 4 a 5 opakujte, dokud nedojde k vyprázdnění lahvičky.
-
7. Sejměte lahvičku aplikační sady CERNEVIT BIO-SET a zlikvidujte ji.
-
8. Lehce promíchejte.
Vícekomorové vaky:
Rozpuštění přípravku CERNEVIT BIO-SET je nutno provést před aktivací vícekomorového vaku (před rozpojením švů a před smícháním obsahu jednotlivých komor).
-
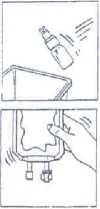
1. Položte vícekomorový vak na vodorovnou plochu.
-
2. Sejměte uzávěr aplikační sady CERNEVIT BIO-SET pootočením a následným tahem za jisticí pruh uzávěru.
-
3. Aplikační sadu BIO-SET napojte na injekční vstup vícekomorového vaku.
-
4. Tlakem na průhlednou pohyblivou část aplikační sady propíchněte pryžový uzávěr lahvičky.
-
5. Lahvičku držte ve vertikální poloze. Několikrát lehce stlačte vak, čímž naplníte lahvičku (přibližně 5 ml) roztokem. Protřepáním lahvičky dojde k rozpuštění přípravku CERNEVIT.
-
6. Propojený vak a aplikační sadu obraťte vzhůru nohama. Několikrát lehce stlačte vak, čímž dojde k vytlačení vzduchu do lahvičky a roztoku do vaku.
-
7. Body 5 a 6 opakujte, dokud nedojde k vyprázdnění lahvičky.
-
8. Sejměte lahvičku aplikační sady CERNEVIT BIO-SET a zlikvidujte ji.
-
9. Nakonec aktivujte vícekomorový vak.
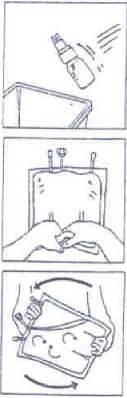
-
10. Otáčením celého vaku (nejméně 3×) důkladně promíchejte.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
BAXTER CZECH spol. s r.o.
Karla Engliše 3201/6, 150 00 Praha 5
Česká republika
8. REGISTRAČNÍ ČÍSLO
76/296/96-C
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22.5.1996
Datum posledního prodloužení registrace: 8.4.2015
Další informace o léčivu CERNEVIT
Jak
se CERNEVIT
podává: intravenózní podání - prášek pro injekční/infuzní roztok
Výdej
léku: na lékařský předpis
Balení: Injekční lahvička
Velikost
balení: 10 SET
Držitel rozhodnutí o registraci daného léku v České republice:
BAXTER CZECH spol. s r.o., Praha
E-mail: magdalena_brunova@baxter.com; dana_sladeckova@baxter.com
Telefon: 225774141, 225774143







