Souhrnné informace o léku - VONILLE 0,060 MG/0,015 MG POTAHOVANÉ TABLETY
1.
Vonille 0,060 mg/0,015 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Žluté tablety (aktivní tablety):
Gestodenum 60 mikrogramů
Ethinylestradiolum: 15 mikrogramů
Pomocné látky se známým účinkem: monohydrát laktózy 57,61, sójový lecithin 0,042 mg
Bílé tablety neobsahují léčivé látky (tablety placeba)
Pomocné látky: monohydrát laktózy 70,897 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Každá aktivní tableta je kulatá, hladká, žlutá a potahovaná tableta o průměru 5,5 mm.
Každá tableta placeba je bílá, kulatá a bikonvexní tableta o průměru 5,5 mm.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Perorální antikoncepce.
Rozhodnutí předepsat přípravek Vonille by mělo být provedeno po zvážení jednotlivých současných rizikových faktorů ženy, zvláště rizikových faktorů pro žilní tromboembolismus (VTE), a toho, jaké je riziko VTE u přípravku Vonille v porovnání s dalšími přípravky CHC (viz body 4.3 a 4.4).
4.2 Dávkování a způsob podání
Cesta podání: perorální podání
Tablety se musí užívat každý den přibližně ve stejnou dobu, v případě potřeby s trochou tekutiny, v pořadí uvedeném na obalu blistru. Užívání tablet je trvalé. Užívá se jedna tableta denně po dobu 28 po sobě jdoucích dnů. Každý následný blistr se začne užívat den po užití poslední tablety z předchozího blistru. Krvácení po vysazení obvykle začíná druhý až třetí den po zahájení užívání tablet placeba (poslední řada) a nemusí skončit před zahájením užívání tablet z dalšího blistru.
- Nepředcházelo-li užívání hormonální antikoncepce [v předchozím měsíci]
- Přechod z kombinované hormonální antikoncepce (kombinovaná perorální antikoncepce (COC), vaginální kroužek nebo transdermální náplast)
- Přechod z antikoncepční metody zahrnující pouze progestagen (tableta obsahující pouze progestagen, injekce, implantát) nebo z nitroděložního systému uvolňujícího progestagen (IUS).
- Užívání po potratu v prvním trimestru
- Užívání po porodu nebo potratu v druhém trimestru
Placebo tablet z poslední (4.) řady blistru si nemusí všímat. Naopak, měla by vynechané placebo tablety zlikvidovat pro zabránění neúmyslnému prodloužení fáze užívání tablet placeba. Následující rada se týká pouze vynechání aktivních tablet:
Pokud se užití tablety opozdí o méně než 12 hodin, není antikoncepční ochrana narušena. Žena by měla užít tabletu okamžitě, jakmile si chybu uvědomí a další tabletu pak užije v obvyklou dobu.
Pokud se užití tablety opozdí o více než 12 hodin, může být antikoncepční ochrana snížena. Další opatření se pak mohou řídit následujícími dvěma základními pravidly:
-
1. Užívání tablet nesmí být nikdy přerušeno na dobu delší než 4 dny.
-
2. K dosažení odpovídající suprese hypothalamo-hypofyzo-ovariální osy je třeba 7 dnů nepřerušeného užívání tablet.
-
V souladu s těmito pravidly lze v běžné praxi poskytnout následující doporučení:
Uživatelka musí užít poslední vynechanou tabletu okamžitě, jakmile si chybu uvědomí, i kdyby to znamenalo užití dvou tablet současně. Poté pokračuje v užívání tablet v obvyklou dobu. Navíc je třeba používat v následujících 7 dnech bariérovou metodu antikoncepce jako např. kondom. Pokud došlo v předchozích 7 dnech k pohlavnímu styku, je třeba uvážit možnost otěhotnění. Čím více tablet bylo vynecháno a čím blíže byly tyto tablety k pravidelnému intervalu bez užívání, tím větší je riziko otěhotnění.
8. – 14. denUživatelka musí užít poslední vynechanou tabletu okamžitě, jakmile si chybu uvědomí, i kdyby to znamenalo užití dvou tablet současně. Poté pokračuje v užívání tablet v obvyklou dobu. Pokud žena užívala tablety pravidelně po dobu 7 dnů před první vynechanou tabletou, další antikoncepční opatření nejsou nutná. Není-li tomu tak, nebo vynechala-li žena více než 1 tabletu, je třeba doporučit zvláštní antikoncepční opatření po dobu 7 dnů.
15. – 24. denVzhledem k nadcházejícímu intervalu bez užívání tablet je velké nebezpečí snížení spolehlivosti antikoncepce. Přesto však upravením schématu užívání lze předejít snížení antikoncepční ochrany. Bude-li se pacientka řídit některým z následujících dvou možných postupů, není třeba používat další antikoncepční opatření za předpokladu, že po dobu 7 dnů předcházejících vynechání první tablety užila všechny tablety správně. Není-li tomu tak, žena musí zvolit první z následujících dvou možností a použít navíc další antikoncepční opatření po dobu 7 dnů.
-
1. Uživatelka musí užít poslední vynechanou tabletu okamžitě, jakmile si chybu uvědomí, i kdyby to znamenalo užití dvou tablet současně. Poté pokračuje v užívání tablet v obvyklou dobu. 4 placebo tablety z poslední řady musí být zlikvidovány. Užívání z následujícího blistru pak zahájí okamžitě. Krvácení z vysazení se pravděpodobně dostaví až po využívání druhého blistru, ale během užívání tablet může dojít ke špinění nebo krvácení z průniku.
-
2. Ženě lze také poradit, aby přerušila užívání tablet ze stávajícího blistru. Tím vznikne interval 4 dnů bez užívání tablet včetně dnů, kdy byly tablety vynechány, a následuje užívání z dalšího blistru.
Pokud žena zapomene užít tablety a následně se nedostaví krvácení z vysazení v prvním normálním intervalu bez užívání tablet, je třeba zvážit možnost těhotenství.
-
V případě závažnějších gastrointestinálních obtíží (zvracení, průjem) nemusí dojít k úplnému vstřebání a je třeba dalších antikoncepčních opatření. Dojde-li během 3 – 4 hodin po užití tablety ke zvracení, měla by se co nejdříve užít nová (náhradní) tableta. Nová tableta by se měla pokud možno užít do 12 hodin od obvyklé doby užití tablety. Pokud uplynulo více než 12 hodin, platí doporučení pro vynechané tablety uvedené v bodě 4.2 „Postup při vynechání tablet“, je-li použitelné. Pokud žena nechce přejít z normálního plánu užívání tablet, musí užít zvláštní tabletu/y z jiného blistru.
Přeje-li si žena oddálit krvácení, musí pokračovat v užívání tablet z dalšího blistru přípravku Vonille bez užívání placebo tablet ze současného blistru. Tak lze pokračovat podle potřeby až do využívání aktivních tablet z druhého blistru. Během této doby může žena pozorovat krvácení z průniku nebo špinění. Po fázi tablet placeba pak žena opět pokračuje v pravidelném užívání přípravku Vonille.
Přeje-li si žena přesunout periodu na jiný den v týdnu, než na který vychází ve stávajícím schématu užívání, lze jí doporučit, aby zkrátila nastávající interval bez užívání tablet o tolik dnů, o kolik si přeje. Čím kratší bude interval, tím větší je riziko, že nedojde ke krvácení z vysazení, ale že bude docházet během užívání z následujícího blistru ke krvácení z průniku a špinění (podobně jako při oddálení periody).
4.3 Kontraindikace
Kombinovaná hormonální antikoncepce (CHC) by se neměla používat u následujících stavů. Pokud se některý z těchto stavů objeví poprvé v průběhu užívání kombinovaného perorálního kontraceptiva, užívání přípravku je třeba okamžitě ukončit.
- Přítomnost nebo riziko žilního tromboembolismu (VTE)
o žilní tromboembolismus – současný žilní tromboembolismus (léčený pomocí antikoagulancií) nebo anamnéza VTE (např. hluboká žilní trombóza [DVT] nebo plicní embolie [PE])
o známá dědičná nebo získaná predispozice pro žilní tromboembolismus, jako je rezistence na APC (včetně faktoru V Leiden), deficit antitrombinu III, deficit proteinu C, deficit proteinu S
o velký chirurgický zákrok s déletrvající imobilizací (viz bod 4.4)
o vysoké riziko žilního tromboembolismu v důsledku přítomnosti více rizikových faktorů (viz bod 4.4);
- Přítomnost nebo riziko arteriálního tromboembolismu (ATE)
o arteriální tromboembolismus – současný arteriální tromboembolismus, anamnéza arteriálního tromboembolismu (např. infarkt myokardu) nebo prodromální stav (např. angina pectoris);
o cerebrovaskulární onemocnění – současná cévní mozková příhoda, anamnéza cévní mozkové příhody nebo prodromálního stavu (např. tranzitorní ischemická ataka, TIA);
o známá hereditární nebo získaná predispozice k arteriálnímu tromboembolismu, jako je hyperhomocysteinémie a antifosfolipidové protilátky (antikardiolipinové protilátky, lupus antikoagulans);
o anamnéza migrény s fokálními neurologickými příznaky;
o vysoké riziko arteriálního tromboembolismu v důsledku vícečetných rizikových faktorů (viz bod 4.4) nebo přítomnosti jednoho závažného rizikového faktoru, jako je:
-
■ diabetes mellitus s cévními příznaky;
-
■ závažná hypertenze;
-
■ závažná dyslipoproteinémie.
- Pankreatitida nebo její anamnéza, pokud souvisela se závažnou hypertriglyceridémií
- Přítomnost nebo anamnéza závažného onemocnění jater až do navrácení jaterních funkcí k normálu.
- Přítomnost nebo anamnéza jaterních nádorů (benigních nebo maligních).
- Známé nebo suspektní malignity ovlivnitelné pohlavními hormony (např. genitální orgány či prsa).
- Nediagnostikované vaginální krvácení
- Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1
- Pacienti s alergií na sóju
Užívání přípravku Vonille je kontraindikováno při současném užívání léčivých přípravků obsahujících ombitasvir/paritaprevir/ritonavir a dasabuvir (viz body 4.4 a 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Obecná
Pokud jsou přítomna jakákoli onemocnění nebo rizikové faktory uvedené níže, měla by být vhodnost přípravku Vonille s ženou prodiskutována.
-
V případě zhoršení nebo prvního výskytu jakéhokoli z těchto stavů nebo rizikových faktorů by mělo být ženě doporučeno, aby kontaktovala svého lékaře, který stanoví, zda by měla užívání přípravku Vonille ukončit.
-
V případě podezření nebo prokázaného výskytu VTE nebo ATE je potřeba podávání CHC přerušit.
-
V případě nasazení antikoagulační léčby je potřeba zahájit podávání vhodné alternativní antikoncepce vzhledem k teratogenitě antikoagulační léčby (kumariny).
-
1. Riziko žilního tromboembolismu (VTE)
Užívání jakékoli kombinované hormonální antikoncepce (CHC) zvyšuje riziko žilního tromboembolismu (VTE) ve srovnání s jejím neužíváním. Přípravky, které obsahují levonorgestrel, norgestimát nebo norethisteron jsou spojovány s nejnižším rizikem VTE. Další přípravky, jako je přípravek Vonille mohou mít až dvakrát vyšší úroveň rizika. Rozhodnutí používat jakýkoli přípravek jiný než ten, který má nejnižší riziko VTE, by mělo být učiněno po diskusi se ženou, aby se zajistilo, že rozumí riziku VTE u přípravku Vonille, rozumí, jak její současné rizikové faktory toto riziko ovlivňují a že riziko VTE je nejvyšší v prvním roce užívání léku. Existují také některé důkazy, že riziko je zvýšené, když je CHC opětovně zahájena po pauze v užívání trvající 4 týdny nebo déle.
U žen, které neužívají CHC a nejsou těhotné, se asi u 2 z 10 000 vyvine VTE v průběhu jednoho roku. U každé jednotlivé ženy však může být riziko daleko vyšší v závislosti na jejích základních rizikových faktorech (viz níže).
Odhaduje se1 , že z 10 000 žen, které používají CHC obsahující gestoden se u 9 až 12 žen vyvine VTE během jednoho roku; v porovnání s přibližně 62 případy u žen, které používají CHC obsahující levonorgestrel.
VTE může být fatální v 1–2 % případů.
-
1 Tyto incidence byly odhadnuty ze souhrnu dat z epidemiologických studií s použitím relativních rizik pro různé přípravky ve srovnání s CHC obsahující levonorgestrel.
-
2 Střední bod rozmezí 5–7 na 10 000 WY (žen-roků) na základě relativního rizika pro CHC obsahující levonorgestrel oproti jejímu nepoužívání přibližně 2,3 až 3,6
Počet příhod
VTE
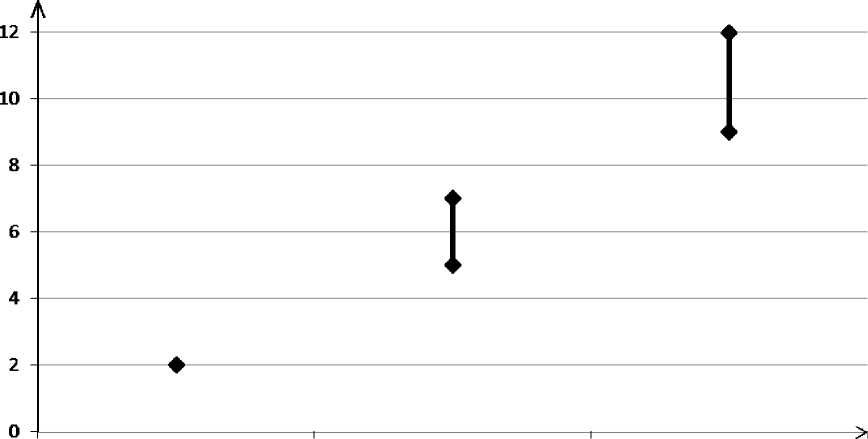
Neuživatelka CHC (2 příhody) i CHC obsahující levonorgestrel ; CHC obsahující gestoden
(5–7 příhod) (9–12 příhod)
Extrémně vzácně byla hlášena trombóza u uživatelek CHC v dalších cévách, např. jaterních, mezenterických, renálních nebo retinálních žilách a tepnách.
Riziko žilních tromboembolických komplikací u uživatelek CHC se může podstatně zvyšovat u ženy, která má další rizikové faktory, zvláště pokud je přítomno více rizikových faktorů (viz tabulka). Přípravek Vonille je kontraindikován, pokud má žena více rizikových faktorů, které pro ni představují vysoké riziko žilní trombózy (viz bod 4.3). Pokud má žena více než jeden rizikový faktor, je možné, že zvýšení rizika je vyšší než součet jednotlivých faktorů – v tomto případě by mělo být zváženo její celkové riziko VTE. Pokud je poměr přínosů a rizik považován za negativní, neměla by být CHC předepisována (viz bod 4.3).
Tabulka: Rizikové faktory VTE
| Rizikový faktor | Poznámka |
| Obezita (index tělesné hmotnosti nad 30 kg/m2) | Při zvýšení BMI se významně zvyšuje riziko. Zvláště důležité je zvážit, zda jsou také přítomny další rizikové faktory. |
| Prodloužená imobilizace, velký | V těchto situacích je doporučeno ukončit |
| chirurgický zákrok, jakýkoli chirurgický zákrok na nohách a pánvi, neurochirurgický zákrok nebo větší trauma. Poznámka: dočasná imobilizace, včetně cestování letadlem > 4 hodiny může být také rizikovým faktorem VTE, zvláště u žen s dalšími rizikovými faktory | používání/užívání náplasti/pilulky/kroužku (v případě plánovaného chirurgického výkonu minimálně 4 týdny předem) a nezahajovat užívání/používání do dvou týdnů po kompletní remobilizaci. Měla by se použít další antikoncepční metoda pro zabránění nechtěnému těhotenství. Antitrombotická léčba by měla být zvážena, pokud přípravek Vonille nebyl předem vysazen. |
| Pozitivní rodinná anamnéza (žilní tromboembolismus kdykoli u sourozence nebo rodiče, zvláště v relativně nízkém věku např. do 50 let věku). | Pokud je suspektní hereditární predispozice, měla by být žena před rozhodnutím o používání jakékoli CHC odeslána k odborníkovi na konzultaci |
| Další onemocnění související s VTE | Zhoubné onemocnění, systémový lupus erytematodes, hemolyticko-uremický syndrom, chronické zánětlivé onemocnění střev (Crohnova choroba nebo ulcerózní kolitida) a srpkovitá anémie |
| Vyšší věk | Zvláště nad 35 let |
Není žádná shoda o možné roli varixů a povrchové tromboflebitidy v nástupu nebo progresi žilní trombózy.
Zvýšené riziko tromboembolismu v těhotenství a zvláště během šestinedělí musí být zváženo (pro informaci o „Fertilita, těhotenství a kojení “ viz bod 4.6).
V případě příznaků by mělo být ženě doporučeno, aby vyhledala naléhavou lékařskou péči a informovala lékaře, že užívá CHC.
Příznaky hluboké žilní trombózy (DVT) mohou zahrnovat:
-
– jednostranný otok nohy a/nebo chodidla nebo podél žíly v noze;
-
– bolest nebo citlivost v noze, která může být pociťována pouze vstoje nebo při chůzi;
-
– zvýšenou teplotu postižené nohy, zarudnutí nebo změnu barvy kůže nohy.
-
– náhlý nástup nevysvětlitelné dušnosti nebo rychlého dýchání;
-
– náhlý kašel, který může být spojený s hemoptýzou;
-
– ostrou bolest na hrudi;
-
– těžké točení hlavy nebo závrať způsobené světlem;
-
– rychlý nebo nepravidelný srdeční tep.
-
2. Riziko arteriálního tromboembolismu (ATE)
Epidemiologické studie spojovaly používání CHC se zvýšeným rizikem arteriálního tromboembolismu (infarkt myokardu) nebo cerebrovaskulární příhody (např. tranzitorní ischemická ataka, cévní mozková příhoda). Arteriální tromboembolické příhody mohou být fatální.
Riziko arteriálních tromboembolických komplikací nebo cerebrovaskulární příhody u uživatelek CHC se zvyšuje u žen s rizikovými faktory (viz tabulka). Přípravek Vonille je kontraindikován, pokud má žena jeden závažný rizikový faktor nebo více rizikových faktorů ATE, které pro ni představují riziko arteriální trombózy (viz bod 4.3). Pokud má žena více než jeden rizikový faktor, je možné, že zvýšení rizika je vyšší než součet jednotlivých faktorů – v tomto případě by mělo být zváženo její celkové riziko. Pokud je poměr přínosů a rizik považován za negativní, neměla by být CHC předepisována (viz bod 4.3).
Tabulka: Rizikové faktory ATE
| Rizikový faktor | Poznámka |
| Vyšší věk | Zvláště nad 35 let |
| Kouření | Ženě by mělo být doporučeno, aby nekouřila, pokud chce používat CHC. Ženám ve věku nad 35 let, které dále kouří, by mělo být důrazně doporučeno, aby používaly jinou metodu antikoncepce. |
| Hypertenze | |
| Obezita (index tělesné hmotnosti nad 30 kg/m2) | Při zvýšení BMI se významně zvyšuje riziko. Zvláště důležité u žen s dalšími rizikovými faktory |
| Pozitivní rodinná anamnéza (arteriální tromboembolismus kdykoli u sourozence nebo rodiče, zvláště v relativně nízkém věku např. do 50 let věku). | Pokud je suspektní hereditární predispozice, měla by být žena odeslána k odborníkovi na konzultaci před rozhodnutím o používání jakékoli CHC |
| Migréna | Zvýšení frekvence nebo závažnosti migrény během používání CHC (což může být prodromální známka cévní mozkové příhody) může být důvodem okamžitého ukončení léčby |
| Další onemocnění související s nežádoucími cévními příhodami | Diabetes mellitus, hyperhomocysteinémie, chlopenní srdeční vada a fibrilace síní, dyslipoproteinémie a systémový lupus erytematodes. |
V případě příznaků by mělo být ženě doporučeno, aby vyhledala naléhavou lékařskou péči a informovala lékaře, že užívá CHC.
Příznaky cévní mozkové příhody mohou zahrnovat:
-
– náhlou necitlivost nebo slabost obličeje, paže nebo nohy, zvláště na jedné straně těla;
-
– náhlé potíže s chůzí, závratě, ztrátu rovnováhy nebo koordinace;
-
– náhlou zmatenost, problémy s řečí nebo porozuměním;
-
– náhlé potíže se zrakem na jednom nebo obou očích;
-
– náhlou, závažnou nebo prodlouženou bolest hlavy neznámé příčiny;
-
– ztrátu vědomí nebo omdlení s nebo bez záchvatu.
-
– bolest, nepříjemný pocit, tlak, těžkost, pocit stlačení nebo plnosti na hrudi, v paži nebo pod hrudní kostí;
-
– nepříjemný pocit vyzařující do zad, čelisti, hrdla, paže, žaludku;
-
– pocit plnosti, poruchu trávení nebo dušení;
-
– pocení, nauzeu, zvracení nebo závratě;
-
– extrémní slabost, úzkost nebo dušnost;
-
– rychlý nebo nepravidelný srdeční tep.
Před prvním zahájením nebo znovuzahájením léčby přípravkem Vonille by měla být získána kompletní anamnéza (včetně rodinné anamnézy) a musí být vyloučeno těhotenství. Měl by se změřit krevní tlak a mělo by být provedeno tělesné vyšetření při zvážení kontraindikací (viz bod 4.3) a varování (viz bod 4.4). Je důležité, aby byla žena upozorněna na informace o žilní a arteriální trombóze, včetně rizika přípravku Vonille v porovnání s dalšími typy CHC, na příznaky VTE a ATE, známé rizikové faktory a co by měla dělat v případě suspektní trombózy.
Žena by také měla být informována, aby si pečlivě přečetla příbalovou informaci pro uživatele a aby dodržovala uvedené instrukce. Frekvence a povaha vyšetření by měly být založeny na stanovených postupech a upraveny podle individuálních potřeb ženy.
Ženy by měly být informovány, že hormonální antikoncepce nechrání před HIV infekcí (AIDS) a dalšími sexuálně přenosnými chorobami.
Přítomnost jednoho závažného rizikového faktoru nebo více rizikových faktorů žilní nebo arteriální choroby může také představovat kontraindikaci.
-
3. Nádory
Karcinom děložního hrdla
V některých epidemiologických studiích bylo naznačeno, že dlouhodobé užívání kombinovaných perorálních kontraceptiv (COC) může dále přispívat k tomuto zvýšenému riziku, ale není dosud jasné, do jaké míry může být tento nález ovlivněn dalšími faktory sexuálního chování a dalšími faktory, jako je lidský papilomavirus (HPV).
Karcinom prsu
Meta-analýza z 54 epidemiologických studií hovoří o lehce zvýšeném relativním riziku (RR = 1,24) diagnózy karcinomu prsu u žen, které právě užívají COC. Toto zvýšené riziko postupně klesá během 10 let po ukončení užívání COC. Vzhledem k tomu, že karcinom prsu je vzácný u žen do 40 let, zvýšení počtu diagnostikovaných karcinomů prsu u současných a dřívějších uživatelek COC je malé ve vztahu k celkovému riziku karcinomu prsu.
Tyto studie neposkytují důkaz kauzality. Příčinou pozorovaného zvýšení rizika karcinomu prsu u uživatelek COC může být časnější diagnóza, biologický účinek COC nebo kombinace obojího. Karcinom prsu diagnostikovaný u současných nebo minulých uživatelek bývá klinicky méně pokročilý než u žen, které COC nikdy neužívaly.
Tumory jater
U uživatelek COC byly hlášeny benigní a maligní jaterní tumory. Vzácně byly tyto tumory příčinou život ohrožujícího nitrobřišního krvácení. Objeví-li se silná bolest v nadbřišku, zvětšení jater nebo známky nitrobřišního krvácení u ženy užívající COC, je třeba v diferenciální diagnóze vzít v úvahu možnost hepatálního tumoru.
-
4. Další stavy
U žen, které trpí hypertriglyceridemií, nebo které mají toto onemocnění v rodinné anamnéze, může být v průběhu užívání kombinovaných perorálních kontraceptiv (COC) zvýšené riziko pankreatitidy.
Přerušení COC může být nevyhnutelné při akutních a chronických poruchách jaterních funkcí na dobu, než se markery jaterních funkcí vrátí k normálním hodnotám. Přerušení užívání COC rovněž vyžaduje recidiva cholestatické žloutenky a/nebo cholestatického pruritu, které se objevily v těhotenství nebo během dřívějšího užívání pohlavních steroidů.
Přestože bylo u mnoha žen užívajících COC zaznamenáno lehké zvýšení krevního tlaku, klinicky významný vzestup je vzácný. Pokud se však v průběhu užívání COC rozvine klinicky signifikantní hypertenze, je třeba kombinovanou perorální antikoncepci vysadit a léčit hypertenzi. Uzná-li lékař za vhodné, lze COC opět nasadit, pokud je antihypertenzní terapií dosaženo normálních hodnot krevního tlaku.
O zhoršení nebo prvním projevu následujících stavů se hovoří v souvislosti jak s těhotenstvím tak užíváním COC, ale důkaz souvislosti s užíváním COC není přesvědčivý: žloutenka a/nebo pruritus související s cholestázou, tvorba žlučových kamenů, porfyrie, systémový lupus erythematodes, hemolyticko-uremický syndrom, Sydenhamova chorea, gestační herpes, ztráta sluchu způsobená otosklerózou.
U žen s hereditárním angioedémem mohou estrogeny indukovat nebo exacerbovat příznaky angioedému.
COC mohou mít vliv na periferní inzulínovou rezistenci a glukózovou toleranci. Proto by měly být diabetičky pečlivě sledovány během užívání COC.
Jedna žlutá tableta tohoto léčivého přípravku obsahuje 57,61 mg laktózy, bílá tableta obsahuje 70, 897 mg. Pacientky se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo malabsorpcí glukózy a galaktózy by neměly tento přípravek užívat.
Během užívání COC bylo hlášeno zhoršení endogenní deprese, epilepsie (viz bod 4.5 Interakce), Crohnovy choroby a ulcerózní kolitidy.
Někdy se může objevit chloasma a to zvláště u žen, které mají v anamnéze chloasma gravidarum. Ženy, které mají dispozici ke vzniku chloasmat, by se měly během užívání COC vyhnout slunění a expozici ultrafialovému záření.
Během klinických studií provedených s pacienty léčenými s virovou hepatitidou C (HCV) léčivými přípravky obsahujícími ombitasvir/paritaprevir/ritonavir a dasabuvir s nebo bez ribavirinu se u žen užívajících léčivé přípravky s ethinylestradiolem, jako jsou např. kombinovaná hormonální kontraceptiva (CHC), vyskytlo výrazně častěji zvýšení aminotransferázy (ALT) na více než pětinásobek horní hranice normálních hodnot (ULN) (viz body 4.3 a 4.5).
Účinnost perorálních kontraceptiv může být snížena například při vynechání tablet (bod 4.2), v případě těžkého průjmu nebo zvracení (bod 4.2), nebo při současném užívání dalších léků (bod 4.5)
Při užívání kteréhokoliv kombinovaného perorálního kontraceptiva (COC) se může objevit nepravidelné krvácení (špinění nebo krvácení z průniku) a to především během prvních měsíců užívání. Z toho důvodu má hledání příčiny nepravidelného krvácení smysl až po adaptačním intervalu přibližně tří cyklů.
Pokud nepravidelné krvácení pokračuje nebo se objeví po období pravidelných cyklů, pak je třeba uvážit možnost nehormonální příčiny a provést odpovídající diagnostické kroky k vyloučení malignity nebo těhotenství. Může sem patřit kyretáž.
U některých žen nemusí dojít během intervalu bez užívání tablet ke krvácení z vysazení. Je-li COC užíváno podle pokynů popsaných v bodu 4.2, je nepravděpodobné, že je žena těhotná. Pokud však COC nebylo užíváno před prvním vynechaným krvácením pravidelně nebo nedošlo-li ke krvácení z vysazení dvakrát, je třeba před dalším užíváním COC vyloučit těhotenství.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Pozn.: měly by být konzultovány odborné informace k doprovodné léčbě, aby byly zjištěny možné interakce.
Mohou se objevit interakce s léky, které indukují mikrozomální enzymy, což může mít za následek zvýšenou clearance pohlavních hormonů a může vést ke krvácení z průniku a/nebo k selhání kontracepce.
Postup
Enzymová indukce může být pozorována již po několika dnech léčby. Maximální enzymová indukce je obvykle pozorována během několika týdnů. Po přerušení léčby může enzymová indukce přetrvávat po dobu přibližně 4 týdnů.
Krátkodobá léčba
Ženy, léčené léčivými látkami indukujícími enzymy by měly dočasně používat navíc k COC bariérovou metodu kontracepce nebo jiný způsob kontracepce. Po celou dobu léčby souběžně podávaným přípravkem a následujících 28 dnů po ukončení léčby musí být používána bariérová metoda kontracepce.
Pokud léčba zasáhne do období ukončení užívání aktivních tablet COC ze stávajícího blistru, placebo tablety musí být vyřazeny a ihned má být zahájeno užívání dalšího blistru COC.
Dlouhodobá léčba
Pokud je žena na dlouhodobé léčbě léčivými látkami indukujícími enzymy, doporučuje se používat jinou spolehlivou nehormonální metodu kontracepce.
Látky, které zvyšují clearance COC (snižující účinky COC enzymovou indukcí), např.:
Barbituráty, bosentan, karbamazepin, fenytoin, primidon, rifampicin a léky na HIV – ritonavir, nevirapin a efavirenz a pravděpodobně také felbamát, griseofulvin, oxkarbazepin, topiramát a přípravky obsahující třezalku tečkovanou (Hypericum perforatum).
Léčivé přípravky zvyšující gastrointestinální motilitu, např. metoklopramid, mohou snižovat plazmatické koncentrace přípravku Vonille.
Farmakodynamické interakce
Současné podávání s léčivými přípravky obsahujícími ombitasvir/paritaprevir/ritonavir a dasabuvir s nebo bez ribavirinu může zvýšit riziko zvýšení hladiny ALT (viz body 4.3 a 4.4). Proto musí uživatelky přípravku Vonille přejít před zahájením výše uvedené léčby na alternativní antikoncepční metodu (např. kontraceptiva obsahující pouze progestagen, nebo nehormonální metody antikoncepce). Užívání přípravku Vonille může být obnoveno 2 týdny po ukončení léčby tímto kombinovaným režimem.
Látky s variabilními účinky na clearance COC
Při současném užívání s COC, mnoho kombinací inhibitorů HIV proteázy a nenukleosidových inhibitorů reverzní transkriptázy, včetně kombinací s HCV inhibitory může zvyšovat nebo snižovat plazmatické koncentrace estrogenu nebo progestinů. V některých případech může být účinek těchto změn klinicky významný.
Proto je třeba prostudovat informace o přípravku k souběžné léčbě HIV/HCV, aby byly identifikovány potenciální lékové interakce a příslušná doporučení. V případě jakýchkoliv pochybností by navíc ženy léčené inhibitory proteázy nebo nenukleosidovými inhibitory reverzní transkriptázy měly používat bariérovou metodu kontracepce.
- Vliv přípravku Vonille na další léčivé přípravky
- Laboratorní testy
Užívání antikoncepčních steroidů může ovlivnit výsledky některých laboratorních testů, včetně biochemických parametrů jaterních, tyreoidálních, adrenálních a renálních funkcí, plasmatických hladin proteinů (vazebných) např. globulin vážící kortikosteroid a lipidové/lipoproteinové frakce, parametry metabolismu uhlovodanů a parametry koagulace a fibrinolýzy. Změny však obvykle zůstávají v rozmezí normálních laboratorních hodnot.
4.6 Fertilita, těhotenství a kojení
Vonille 0,060 mg/0,015 mg potahované tablety není indikovaný v těhotenství.
Pokud žena otěhotní během užívání přípravku Vonille tablety, musí být další užívání ihned přerušeno.
Rozsáhlé epidemiologické studie však nezaznamenaly zvýšené riziko vrozených vad u dětí narozených ženám užívajícím kombinovaná perorální kontraceptiva před otěhotněním, ani teratogenní vliv kombinovaných perorálních kontraceptiv nezáměrně užívaných v časném těhotenství.
Laktace může být ovlivněna kombinovanými perorálními kontraceptivy, která mohou snižovat množství a měnit složení mateřského mléka. Z toho důvodu se užívání kombinovaných perorálních kontraceptiv obecně nedoporučuje, dokud matka dítě zcela neodstaví. Malé množství antikoncepčních steroidů a/nebo jejich metabolitů může být vylučováno do mléka. Tato množství mohou ovlivnit zdraví dítěte.
Zvýšené riziko VTE během poporodního období je třeba brát v úvahu při znovuzahájení užívání přípravku Vonille (viz bod 4.2 a 4.4).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vonille nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Výskyt amenorhey byl udáván u 15 % žen během klinické studie, viz bod 4.4.
Některé nejčastěji hlášené (víc než 10 %) nežádoucí příhody během studií fáze III a v poregistračním sledování u žen užívajících Vonille jsou bolest hlavy, včetně migrén, náhlé krvácení/špinění.
Další nežádoucí účinky byly hlášeny u žen užívající COC:
| Časté >1 % a <10 % | Méně časté > 0,1% a < 1 % | Vzácné >0,01 % a <0,1 % | Velmi vzácné <0,01 % | |
| Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) | Hepatocelulární karcinom a benigní nádory jater (např. fokální nodulární hyperplazie, hepatální adenom) | |||
| Infekce a infestace | Vaginitida, včetně kandidózy | |||
| Poruchy imunitního systému | Anafylaktické/ anafylaktoidní reakce, včetně velmi vzácných případů kopřivky, angioedému a závažných reakcí s respiračními a cirkulačními příznaky. | Exacerbace systémového lupus erytematodes | ||
| Poruchy metabolismu a | Změny chuti k jídlu (zvýšení | Intolerance glukózy | Exacerbace porfyrie |
| výživy | nebo snížení) | |||
| Psychiatrické poruchy | Změny nálady, včetně deprese, změny libida | |||
| Poruchy nervového systému | Nervozita, závratě | Exacerbace chorey | ||
| Poruchy oka | Intolerance kontaktních čoček | Optická neuritida, retinální vaskulární trombóza | ||
| Gastrointestinální poruchy | Nevolnost, zvracení, abdominální bolest | Abdominální křeče, nadýmání | Pankreatitida | |
| Poruchy jater a žlučových cest | Cholestatická žloutenka | Biliární litiáza a cholestáza1, hepatální a hepatobiliární poruchy (např. hepatidia, abnormální funkce jater) | ||
| Poruchy kůže a podkožní tkáně | Akné | Vyrážka, chloasma (melasma), které mohou přetrvávat, hirsutismus, alopecie. | Erythema nodosum | Erythema multiforme |
| Poruchy ledvin a močových cest | Hemolyticko-uremický syndrom | |||
| Poruchy reprodukčního systému a prsu | Bolest prsů, citlivost, zvětšení, sekrece, dysmenorhea, změna menstruačního cyklu, změna cervikálního ektropie a sekrece. | |||
| Celkové poruchy a reakce v místě aplikace | Retence tekutin/edém | |||
| Cévní poruchy | Zvýšení krevního tlaku | Žilní tromboembolismus (VTE), arteriální tromboembolismus (ATE) | ||
| Vyšetření | Změny tělesné hmotnosti (zvýšení nebo snížení) | Změny hladiny lipidů v séru, včetně hypertriglyceridé mie |
1CHC mohou zhoršovat stávající biliární litiázu a cholestázu
Popis vybraných nežádoucích účinků
U žen užívajících CHC bylo pozorováno zvýšené riziko arteriálních a žilních trombotických a tromboembolických příhod, včetně infarktu myokardu, cévní mozkové příhody, tranzitorních ischemických atak, žilní trombózy a plicní embolie a je podrobněji popsáno v bodě 4.4.
Následující závažné nežádoucí účinky byly hlášeny u žen užívajících COC, které jsou uvedeny v bodu
4.4 Zvláštní upozornění a opatření pro použití
- Hypertenze;
- Tumory jater;
- Výskyt nebo zhoršení stavů, pro které souvislost s COC není přesvědčivá jako např. Crohnova choroba, ulcerózní kolitida, epilepsie, migréna, děložní myom, porfyrie, systémový lupus erytematodes, gestační herpes, Sydenhamova chorea, hemolyticko-uremický syndrom, cholestatická žloutenka;
- Chloasma;
- Přerušení COC může být nevyhnutelné při akutních a chronických poruchách jaterních funkcí na dobu, než se markery jaterních funkcí vrátí k normálním hodnotám.
- U žen s hereditárním angioedémem mohou exogenní estrogeny indukovat nebo exacerbovat příznaky angioedému.
Frekvence diagnózy karcinomu prsu se velmi mírně zvyšuje u uživatelek COC. Karcinom prsu je vzácný u žen do 40 let věku a zvýšený počet je malý v poměru k celkovému riziku karcinomu prsu. Kauzální vztah k COC není znám. Pro další informace viz body 4.3 a 4.4.
Interakce
Interakce jiných léčivých látek (enzymových induktorů) s perorální antikoncepcí mohou vést ke krvácení z průniku a/nebo k selhání kontracepce (viz bod 4.5).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím:
Státního ústavu pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
Nejsou žádné zprávy o vážných škodlivých účincích z předávkování. Vyskytnout se mohou příznaky jako je nevolnost, zvracení a u mladých dívek slabé vaginální krvácení. Neexistují žádná antidota a další léčba by měla být symptomatická.
5. FARMAKOLOGICKÉ VLASTNOSTI
Farmakoterapeutická skupina (ATC): Hormonální antikoncepce pro systémové použití, progestageny a estrogeny, fixní kombinace
ATC kód: G03AA10
5.1 Farmakodynamické vlastnosti
Celkový index Pearl (těhotenství v důsledku selhání metody + těhotenství v důsledku selhání pacienta) pro ethinylestradiol/gestoden 15/60 ^g je 0,24 (95%CI 0,04–0,57).
Antikoncepční účinek kombinovaných perorálních kontraceptiv (COC) je založen na spolupůsobení různých faktorů. Nejdůležitější z nich je inhibice ovulace a změny endometria.
5.2 Farmakokinetické vlastnosti
Absorpce
Perorálně podaný ethinylestradiol je rychle a kompletně absorbován. Nejvyšší sérové koncentrace okolo 30 pg/ml je dosaženo během 1–1,5 hodiny po podání. Ethinylestradiol podléhá rozšířenému prvnímu průchodu játry, což se projevuje velkými interindividuálními rozdíly. Biologická dostupnost je asi 45 %.
Distribuce
Ethinylestradiol má zdánlivý distribuční objem 15 l/kg a vazba na plasmatické proteiny je asi 98 %. Ethinylestradiol indukuje hepatální syntézu globulinů vážících pohlavní hormony (SHBG) a kortikoidy (CBG). Během léčby dávkou 15 ^g ethinylestradiolu se plasmatická koncentrace SHBG zvyšuje z 86 asi na 200 nmol/l.
Biotransformace
Ethinylestradiol se kompletně metabolizuje (plasmatická clearance je asi 10 ml/min/kg). Metabolity jsou vylučovány močí (40 %) a stolicí (60 %).
Eliminace
Eliminační poločas ethinylestradiolu je asi 15 hodin. Ethinylestradiol se nevylučuje v nezměněné formě v žádném významném rozsahu. Metabolity ethinylestradiolu jsou vylučovány močí a žlučí v poměru 4:6.
Rovnovážný stav
Rovnovážný stav je dosažený během druhé poloviny léčebného cyklu a sérové hladiny ethinylestradiolu se akumulují faktorem asi 1,4 až 2,1.
Absorpce
Perorálně podaný gestoden je rychle a kompletně absorbován. Absolutní biologická dostupnost je 100 %. Po perorální aplikaci jedné dávky 60 ^g gestodenu je dosaženo maximálních plasmatických koncentrací 2 ng/ml asi za 60 minut. Plasmatické koncentrace jsou silně závislé na koncentraci SHBG.
Distribuce
Gestoden má zdánlivý distribuční objem 1,4 l/kg po jedné dávce 60 ^g. Váže se asi z 30 % na plasmatický albumin a z 50 – 70 % na SHBG.
Biotransformace
Gestoden se rozsáhle metabolizuje v procesu metabolismu steroidů. Metabolická clearance je asi 0,8 ml/min/kg po jednorázové dávce 60 ^g. Neúčinné metabolity jsou vyloučeny močí (60 %) a stolicí (40 %).
Eliminace
Zdánlivý eliminační poločas gestodenu je asi 13 hodin. Biologický poločas se prodlužuje na 20 hodin po současném podání ethinylestradiolu.
Rovnovážný stav
Po vícenásobném podání současně s ethinylestradiolem se plasmatické koncentrace zvyšují faktorem 2 – 4.
5.3 Předklinické údaje vztahující se k bezpečnosti
Ethinylestradiol a gestoden nejsou genotoxické. Studie karcinogenity s ethinylestradiolem v monoterapii nebo v kombinaci s různými progestageny neukazují žádné zvláštní karcinogenní riziko pro ženy při užívání v indikaci antikoncepce. Je však třeba uvést, že pohlavní hormony mohou podpořit růst některých hormonálně závislých tkání a nádorů.
Studie reprodukční toxicity u fertility, vývoje plodu nebo reprodukční schopnosti ethinylestradiolu v monoterapii nebo v kombinaci s progestageny neodhalily žádné nežádoucí účinky u člověka při doporučeném podávání.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Vonille 0.060 mg/0.015 mg potahované tablety
Aktivní tablety (žluté tablety):
Monohydrát laktózy
Mikrokrystalická celulóza (E460)
Draselná sůl polakrilinu
Magnesium- stearát (E572)
Potahová vrstva:
Polyvinylalkohol
Oxid titaničitý (E-171)
Sójový lecithin (E322)
Mastek
Žlutý oxid železitý (E-172)
Xanthanová klovatina (E415)
Monohydrát laktózy
Povidon K25 (E1201)
Sodná sůl karboxymethylškrobu (typ A)
Koloidní bezvodý oxid křemičitý (E551)
Oxid hlinitý
Magnesium- stearát (E572)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání. Blistr uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Průhledný až slabě neprůhledný PVC/PVDC-Al blistr.
Velikosti balení:
1×24 žlutých tablet + 4 bílé tablety
3×24 žlutých tablet + 4 bílé tablety
6×24 žlutých tablet + 4 bílé tablety
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Exeltis Czech s.r.o.
Želetavská 1449/9
Praha 4 – Michle
Česká republika
8. REGISTRAČNÍ ČÍSLO
17/263/12-C
Další informace o léčivu VONILLE 0,060 MG/0,015 MG POTAHOVANÉ TABLETY
Jak
se VONILLE 0,060 MG/0,015 MG POTAHOVANÉ TABLETY
podává: perorální podání - potahovaná tableta
Výdej
léku: na lékařský předpis
Balení: Blistr
Velikost
balení: 6X28(24+4)
Držitel rozhodnutí o registraci daného léku v České republice:
Exeltis Czech s.r.o., Praha
E-mail: info.czech@exeltis.com
Telefon: +420 241 480 900