Souhrnné informace o léku - RECOMBINATE 1000 INTERNATIONAL UNIT/5 ML
1. NÁZEV PŘÍPRAVKU
Recombinate 500 International Unit/5 ml prášek a rozpouštědlo pro injekční roztok
Recombinate 1000 International Unit/5 ml prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Octocogum alfa 100 IU v 1 ml rekonstituovaného roztoku
Po rekonstituci: jedna 5 ml lahvička obsahuje octocogum alfa 500 IU
Recombinate 500 International Unit/5 ml obsahuje octocogum alfa 500 IU, rekombinantního koagulačního faktoru VIII, v jedné lahvičce.
Po rekonstituci v 5 ml sterilní vody pro injekci obsahuje přípravek octocogum alfa přibližně 100 IU/ml, rekombinantního koagulačního faktoru VIII.
Octocogum alfa 200 IU v 1 ml rekonstituovaného roztoku
Po rekonstituci: jedna 5 ml lahvička obsahuje octocogum alfa 1000 IU
Recombinate 1000 International Unit/5 ml obsahuje octocogum alfa 1000 IU, rekombinantního koagulačního faktoru VIII, v jedné lahvičce.
Po rekonstituci v 5 ml sterilní vody pro injekci obsahuje přípravek octocogum alfa přibližně 200 IU/ml, rekombinantního koagulačního faktoru VIII.
Účinnost se určuje chromogenním testem dle Evropského lékopisu vzhledem k mega standardu FDA, který je kalibrován vůči standardu WHO. Specifická aktivita přípravku Recombinate je přibližně 40008000 IU/mg proteinu.
Recombinate obsahuje rekombinantní koagulační faktor VIII (INN: octocogum alfa). Octocogum alfa (rekombinantní koagulační faktor VIII) je čištěný protein sestávající z 2332 aminokyselin. Má sekvenci aminokyselin srovnatelnou s faktorem VIII a posttranslační modifikace, které se podobají modifikacím molekuly izolované z plazmy. Rekombinantní koagulační faktor VIII je glykoprotein, který je produkován geneticky modifikovanými savčími buňkami, pocházejícími z buněčné linie z ovarií čínského křečka.
Pomocné látky se známým účinkem: sodík
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý až téměř bílý drobivý prášek. Rozpouštědlo (sterilizovaná voda pro injekci) je čirá a bezbarvá tekutina.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII).
Přípravek neobsahuje von Willebrandův faktor, proto není indikován k léčbě von Willebrandovy choroby. Recombinate je indikován u všech věkových skupin od novorozenců po dospělé.
4.2 Dávkování a způsob podání
Dávkování
Dávkování a délka trvání substituční léčby závisí na závažnosti poruchy hemostázy, na místě a rozsahu krvácení a na klinickém stavu pacienta. Léčba má být prováděna ve spolupráci s lékařem, který má zkušenosti s léčbou poruch hemostázy, a s laboratoří, která má možnost měřit koncentraci antihemofilického faktoru (AHF) v plazmě.
Podávaná dávka faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují k platnému standardu WHO pro přípravky obsahující faktor VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v mezinárodních jednotkách (ve vztahu k mezinárodnímu standardu faktoru VIII v plazmě). Jedna mezinárodní jednotka (IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Očekávaný maximální vzestup hladiny přípravku Recombinate in vivo vyjádřený jako IU/dl plazmy nebo % (procento) normálu může být odhadnut vynásobením podané dávky na kg tělesné hmotnosti (IU/kg) dvěma.
Metoda výpočtu je znázorněna na následujících příkladech.
Předpokládaný vzestup faktoru VIII (%) = počet podaných jednotek x 2% / IU / kg
tělesná hmotnost (kg)
Příklad u dospělého s hmotností 70 kg: 1750 IU x 2% / IU / kg = ~50%
70 kg
nebo
Potřebná dávka (IU): Tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%)
2% / IU / kg
Příklad u dítěte s hmotností 40 kg: 40 kg x 70% = 1400 IU
2% / IU / kg
Pečlivé sledování substituční terapie je důležité zejména při větších chirurgických výkonech nebo život ohrožujícím krvácení. Ačkoli dávkování je možno odhadnout pomocí výše uvedeného výpočtu, důrazně se doporučuje, aby se, kdykoli je to možné, ve vhodných intervalech prováděly odpovídající laboratorní testy včetně opakovaného vyšetření hladiny AHF v plazmě pacienta k ověření dosažení a udržení adekvátních hladin AHF. Jestliže nedojde k dosažení požadovaných hladin AHF v plazmě pacienta nebo jestliže se krvácení nezastaví po odpovídající dávce, je třeba zvážit podezření na přítomnost inhibitoru. Pomocí vhodných laboratorních vyšetření je možno prokázat a kvantifikovat přítomnost inhibitoru na základě mezinárodních jednotek AHF neutralizovaných jedním ml plazmy (Bethesda jednotky) nebo celkovým odhadovaným objemem plazmy. Je-li inhibitor přítomen v titrech nižších než 10 Bethesda jednotek na ml, další podání AHF může inhibitor neutralizovat. Pak by při podání dalších mezinárodních jednotek AHF mělo být dosaženo předpokládané odpovědi. V tomto případě je nezbytná kontrola hladin AHF laboratorními testy. Titry inhibitoru nad 10 Bethesda jednotek na ml mohou znemožnit kontrolu hemostázy pomocí AHF vzhledem k nutnosti podávání velmi vysokých dávek.
Následující dávkovací režim vedený v Tabulce 1 může být použit jako vodítko u dospělých a dětí. Množství a frekvence podávání mají být vždy upraveny dle klinické účinnosti v individuálním případě.
Recombinate může být podáván také k profylaxi krvácení (krátkodobé či dlouhodobé) na základě rozhodnutí lékaře u daného pacienta.
Tab. 1: Dávkovací režim
Krvácení
Stupeň krvácení
Časná hemartróza, krvácení do svalů nebo ústní dutiny
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom
Život ohrožující krvácení, jako např. intrakraniální krvácení, krvácení do krku, těžké abdominální krvácení
Chirurgické výkony
Typ operace
Malý chirurgický výkon včetně extrakce zubu
Požadovaná maximální aktivita AHF v krvi po infuzi (% normálu nebo IU/dl plazmy)
20–40
Frekvence infuzí
30–60
60–100
30–60
Velký chirurgický výkon
80–100
(před operací a po operaci)
Zahajte infuzi každých 12 až 24 hodin po 1–3 dny, dokud krvácení, indikované bolestí nepřejde nebo dokud se nedosáhne vyléčení. Opakujte infuzi každých 12 až 24 hodin, obvykle po 3 dny či více, dokud bolest a nemohoucnost nepřejdou.
Opakujte infuzi každých 8 až 24 hodin, dokud není nebezpečí zažehnáno.
Jediná infuze spolu s perorální antifibrinolytickou léčbou během jedné hodiny je účinná přibližně v 70% případů. Každých 24 hodin, nejméně 1 den, dokud se nedosáhne vyléčení.
Opakujte infuzi každých 8 až 24 hodin podle stavu hojení.
Jedná se o maximální aktivitu AHF u pacientů s očekávaným průměrným poločasem faktoru VIII. Je-li to považováno za nezbytné, maximální aktivita má být stanovena do půl hodiny po podání. U pacientů s relativně krátkým poločasem faktoru VIII může být nutné zvýšení dávky a/nebo frekvence podávání.
Každá lahvička přípravku Recombinate je označena aktivitou antihemofilického faktoru (rekombinantního) vyjádřenou obsahem IU v jedné lahvičce.
Účinnost se vztahuje k mezinárodnímu standardu Světové zdravotnické organizace pro koncentráty faktoru FVIII:C. Pokusy ukázaly, že k získání přesných hladin aktivity má být test účinnosti proveden s použitím plastových zkumavek a pipet a substrátu obsahujícího normální hladiny von Willebrandova faktoru.
K dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu 2 až 3 dnů.
Pacienti mají být sledováni s ohledem na vývoj inhibitorů faktoru VIII. Není-li dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo není-li krvácení kontrolováno vhodnou dávkou, je třeba provést vyšetření na přítomnost inhibitorů faktoru VIII. U pacientů s vysokými hladinami inhibitorů nemusí být léčba faktorem VIII účinná a je třeba zvážit jiné terapeutické možnosti. Léčba takových pacientů má být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií.
Viz také bod 4.4.
Pediatrická populace:
Recombinate je vhodný pro použití u dětí všech věkových skupin, včetně novorozenců (Studie bezpečnosti a účinnosti byly provedenu u dříve léčených i dříve neléčených dětí, viz bod 5.1). Při on-demand léčbě se dávkování u pediatrických pacientů neliší od dospělých pacientů. Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií A mohou být v některých případech nutné kratší intervaly dávkování nebo vyšší dávky než obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu 2–3 dnů.
Způsob podání
Přípravek se podává intravenózně po rekonstituci přiloženým rozpouštědlem (viz bod 6.6). Rekonstituovaný přípravek nesmí být uchováván v chladničce. Doporučuje se podávat Recombinate při pokojové teplotě nejpozději do 3 hodin po rekonstituci. Rychlost podání má být taková, aby bylo zajištěno pohodlí pacienta, maximálně 10 ml/min. Před a během podávání přípravku Recombinate má být měřena tepová frekvence. Dojde-li k signifikantnímu zvýšení tepové frekvence, zpomalením rychlosti podávání nebo dočasným přerušením injekce se obvykle dosáhne rychlého vymizení symptomů (viz body 4.4 a 4.8)
Návod k rekonstituci léčivého přípravku před podáním viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou uvedenou v bodě 6.1. Známá alergická reakce na bovinní, myší či křeččí protein.
4.4 Zvláštní upozornění a opatření pro použití
Byly hlášeny těžké alergické reakce na přípravek Recombinate. Pacienti se známou hypersenzitivitou na myší, bovinní či křeččí protein mají být léčeni s opatrností. Pacienti mají být informováni o časných známkách hypersenzitivních reakcí jako je kopřivka, generalizovaná kopřivka, tlak na hrudi, sípot, hypotenze a anafylaxe. Pokud se objeví alergické nebo anafylaktické reakce, je třeba injekci/infuzi ihned přerušit. Je nutné, aby bylo k dispozici adekvátní vybavení k léčbě šoku.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle imunoglobuliny IgG zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. Riziko vzniku inhibitorů souvisí se závažností onemocnění i s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů, s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze, z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Klinický význam vzniku inhibitorů bude záviset na titru inhibitoru, přičemž inhibitory nízkého titru, které jsou krátkodobě přítomny nebo zůstávají trvale na nízkém titru, představují menší riziko nedostatečné klinické odpovědi než inhibitory vysokého titru.
Obecně platí, že všichni pacienti léčení přípravky s koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být terapie faktorem VIII účinná a je třeba zvážit jiné možnosti léčby. Péče o takové pacienty má být vedena lékaři se zkušenostmi v péči o hemofilii a inhibitory faktoru VIII.
V zájmu pacientů se doporučuje, aby byl, kdykoli je to možné, pokaždé, kdy jim je Recombinate podáván, zaznamenán název a číslo šarže přípravku.
Tento léčivý přípravek obsahuje 1,5 mmol sodíku v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Pediatrická populace:
Zvláštní upozornění a opatření pro použití u pediatrických pacientů se neliší od dospělých pacientů.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
S faktorem VIII nebyly prováděny reprodukční studie u zvířat. Vzhledem ke vzácnému výskytu hemofilie A u žen nejsou zkušenosti s podáváním faktoru VIII během těhotenství a kojení. Proto se má faktor VIII během těhotenství nebo kojení používat pouze tehdy, je-li to nezbytně nutné.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly pozorovány účinky na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Tabulkový souhrn nežádoucích účinků:
Následující tabulka je výčtem nežádoucích účinků ze spontánních hlášení a klinických studií. V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
Frekvence byla vyhodnocena podle následujících kritérií: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000) a velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit).
| Standardní třída orgánových systémů dle MedDRA | Nežádoucí účinek | Četnost |
| Infekce a infestace | Infekce ucha | Méně časté |
| Poruchy krve a lymfatického systému | Inhibice FVIII | Méně časté (PTP) 1 Velmi časté (PUP) 1 |
| Poruchy imunitního systému | Anafylaktická reakce Hypersenzitivita2 | Není známo |
| Poruchy nervového systému | Závrať Třes | Méně časté |
| Ztráta vědomí Synkopa Bolest hlavy Parestezie | Není známo | |
| Srdeční poruchy | Cyanóza Tachykardie | Není známo |
| Cévní poruchy | Epistaxe Zrudnutí Hematom Hypotenze Bledost Periferní chlad | Méně časté |
| Respirační, hrudní a mediastinální poruchy | Faryngolaryngeální bolest | Méně časté |
| Dyspnoe Kašel Sípot | Není známo | |
| Gastrointestinální poruchy | Nauzea | Méně časté |
| Zvracení Bolest břicha | Není známo | |
| Poruchy kůže a podkožní tkáně | Hyperhidróza Svědění Vyrážka Makulopapulární vyrážka | Méně časté |
| Angioedém Kopřivka Kožní exfoliace Erytém | Není známo | |
| Poruchy svalové a kosterní soustavy a pojivové tkáně | Bolest končetin | Méně časté |
| Celkové poruchy a reakce v místě aplikace | Zimnice | Časté |
| Únava Horečka | Méně časté | |
| Malátnost Reakce v místě vpichu Bolest na hrudi Hrudní diskomfort | Není známo | |
| Vyšetření | Abnormální výsledky testů akustické stimulace | Méně časté |
| 1 Četnost vychází ze studií se všemi přípravky s faktorem VIII, které zahrnova | ly pacienty se závažnou | |
hemofilií A. PTP = dříve léčení pacienti, PUP = dříve neléčení pacienti
2 Časné známky hypersenzitivních reakcí jsou např. kopřivka, dyspnoe, kašel, hrudní diskomfort, sípot, anafylaxe, vyrážka, hypotenze, svědění, zimnice, zrudnutí, horečka, cyanóza, tachykardie, zvracení, synkopa, bolest hlavy. Opatrnosti je třeba u pacientů s anamnézou alergických reakcí na složky přípravku (viz body 4.3 a 4.4).
Popis vybraných závažných nežádoucích účinků
K rozvoji neutralizujících protilátek (inhibitorů) může dojít u pacientů s hemofilií A, kteří jsou léčeni faktorem VIII, včetně přípravku Recombinate. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum.
Pediatrická populace
Kromě vývoje inhibitoru u dříve neléčených pacientů (PUPs) nebyly v klinických studiích zaznamenány specifické věkové rozdíly v nežádoucích účincích.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu: Státní ústav pro kontrolu léčiv, Šrobárova 48, 100 41 Praha 10, webové stránky:
4.9 Předávkování
Nejsou známy žádné symptomy předávkování.
5.
FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hemostyptika, hemostatika: krevní koagulační faktor VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktor VIII a von Willebrandův faktor) s různými fyziologickými funkcemi.
Při infuzi pacientovi s hemofilií se faktor VIII váže na von Willebrandův faktor v pacientově krevním oběhu.
Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha srážení krve způsobená sníženými hladinami faktoru VIII:C, což má za následek silné krvácení do kloubů, svalů a vnitřních orgánů, ať už spontánní nebo v důsledku úrazu nebo chirurgického traumatu. Hladiny plazmatického faktoru VIII se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru a úpravu sklonu ke krvácení.
Recombinate byl testován u 71 dosud neléčených dětí. Průměrný věk v této skupině v době první infuze přípravku Recombinate byl 10 měsíců (rozmezí: 2 dny až 50 měsíců). Přípravek byl dobře tolerován a nevyvolával signifikantní krátkodobé nežádoucí účinky. Jeho klinická účinnost byla srovnatelná s jinými molekulami FVIII plné délky v léčbě akutního krvácení i při chirurgické profylaxi (10 pacientů podstoupilo chirurgický zákrok). Při dlouhodobém sledování této skupiny byl zjištěn výskyt nežádoucích účinků souvisejících s přípravkem u 0,86/1000 infuzí, žádný z nich nebyl závažný či život ohrožující.
5.2 Farmakokinetické vlastnosti
Farmakokinetické studie u 69 dříve léčených pacientů zjistily, že průměrný poločas Recombinate v krevním oběhu je 14,6 ± 4,9 hodin (n = 67) a že se statisticky signifikantně neliší od lidského antihemofilického faktoru izolovaného z plazmy – HemofiluM (pdAHF). Průměrný poločas HemofiluM byl 14,7 ± 5,1 hodin (n = 61). Skutečné počáteční recovery pozorované u Recombinate po infuzi dávky 50 IU/kg bylo 123,9 ± 47,7 IU/dl (n = 23), což je signifikantně vyšší než skutečné počáteční recovery u HemofiluM 101,7 ± 31,6 IU/dl (n = 61). Vypočítaný poměr skutečného recovery faktoru VIII vzhledem k předpokládanému recovery (tj. 2% zvýšení aktivity faktoru VIII 1 IU rAHF/kg tělesné hmotnosti) u přípravku Recombinate (121,2 ± 48.9%) je podobný jako u HemofiluM (123,4 ± 16,4%).
Od 68 dosud neléčených pacientů bylo získáno celkem 494 studií recovery faktoru VIII. Dvě stě dvanáct studií recovery faktoru VIII bylo provedeno během léčby pacientů s krvácením s průměrným ± SO skutečným recovery 70,0 ± 37,9 IU/dl (N = 208), čtyři recovery byla z analýzy vynechána z důvodu zkreslení. Tato vysoká variabilita je způsobena širokým rozmezím skutečně podávané dávky (13,8 až
-
103,2 IU/kg (průměr ± SO (standardní odchylka) 36,0 ± 16,2 a střední hladina 30,2 IU/kg). K doložení variabilních hladin dávkování byly vypočítány poměry skutečného recovery faktoru VIII ku předpokládanému recovery s průměrným výsledkem 1,0 ± 0,3.
Celkem 68 studií recovery faktoru VIII bylo získáno během období, kdy pacienti dostávali další infuze k následné léčbě předchozího krvácení. Skutečná hladina recovery faktoru VIII byla korigována na hladinu FVIII před infuzí. Průměr ± SO skutečného recovery faktoru VIII byl 88,6 ± 38,2 IU/dl (N = 66), dvě recovery byla z analýzy vynechána kvůli zkreslení. Široké rozmezí skutečně podávaných dávek, 18,5 až 85,7 IU/kg (průměr ± SO 38,6 ± 15,9 a střední hladina 32,1 IU/kg) opět způsobuje značnou variabilitu pozorovaných hladin recovery. Průměr ± SO skutečného recovery faktoru VIII ku předpokládanému recovery byl 1,0 ± 0,3 se střední hladinou 1,0.
Celkem 214 studií recovery faktoru VIII bylo provedeno u pacientů ve stabilizovaném stavu a bylo zjištěno průměrné skutečné recovery 71,6 ± 29,7 IU/dl (N = 209), pět recovery bylo z analýzy vynecháno kvůli zkreslení. Podávané dávky se pohybovaly od 10,4 do 68,1 IU/kg (průměr ± SO 38,0 ± 12,7 a střední hladina 36,1 IU/kg). Průměr ± SO skutečného recovery faktoru VIII ku předpokládanému recovery byl 1,0 ± 0,3.
5.3 Předklinické údaje vztahující se k bezpečnosti
Recombinate působí jako endogenní faktor VIII. U laboratorních zvířat nebyly zjištěny žádné toxické účinky po podání dávek, které několikanásobně překračovaly doporučenou dávku na kilogram tělesné hmotnosti u člověka. Recombinate byl testován z hlediska mutagenity v dávkách významně překračujících plazmatické koncentrace AHF in vitro a v dávkách až desetinásobně vyšších, než je předpokládaná maximální klinická dávka in vivo, přičemž nebyly zjištěny reverzní mutace, chromozomální aberace či zvýšení mikronukleů v polychromatických erytrocytech kostní dřeně. Jelikož klinické zkušenosti nepřinesly důkazy tumorogenních a mutagenních účinků, dlouhodobé studie u zvířat zaměřené na hodnocení karcinogenního potenciálu nejsou považovány za nutné.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Lidský albumin
Chlorid sodný
Histidin
Makrogol 3350
Dihydrát chloridu vápenatého
Kyselina chlorovodíková (k úpravě pH)
Hydroxid sodný (k úpravě pH)
Rozpouštědlo:
Voda pro injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
Má být použit pouze infuzní set dodávaný s přípravkem, protože v důsledku adsorpce koagulačního faktoru VIII na vnitřní povrchy některých infuzních setů může dojít k selhání léčby.
6.3 Doba použitelnosti
3 roky. Po rekonstituci nemá být přípravek ukládán do chladničky a má být podán do tří hodin.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C – 8°C).
Chraňte před mrazem.
Uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
V rámci doby použitelnosti může být přípravek před použitím uchováván při teplotě 15°C – 25°C až po dobu šesti měsíců.
Po uchovávání při teplotě 15°C – 25°C přípravek nevracejte do chladničky.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Každé balení obsahuje lahvičku s práškem, 5ml lahvičku s rozpouštědlem (obě ze skla třídy I s pryžovými zátkami) a pomůcku pro rekonstituci (BAXJECT II) + jednu sterilní jednorázovou plastovou injekční stříkačku + jeden sterilní mini-infuzní set + 2 dezinfekční polštářky + 2 náplasti.
Alternativně k BAXJECT II může být dodávána sada jehel k rekonstituci sestávající ze sterilní jehly se dvěma konci (k přepuštění rozpouštědla do lahvičky s Recombinate) a jedné sterilní filtrační jehly (k přepuštění rekonstituovaného roztoku do stříkačky).
Velikost balení: 1
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek je určen k intravenóznímu podání po rekonstituci přiloženou sterilní vodou pro injekci. Je nutno použít jednorázovou plastovou injekční stříkačku dodávanou s přípravkem.
-
– Použijte do tří hodin po rekonstituci.
-
– Po rekonstituci přípravek nevracejte do chladničky.
-
– Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
-
– Roztok má být čirý nebo mírně opalescentní. Zakalené roztoky nebo roztoky obsahující částice nepoužívejte. Rekonstituovaný přípravek má být před podáním vizuálně zkontrolován s ohledem na obsah částic nebo změnu zbarvení.
-
– Nepoužívejte, pokud je přípravek, systém jeho sterilní bariéry nebo obal poškozený nebo pokud vykazuje jakékoli známky zhoršené kvality.
Rekonstituce: použijte aseptický postup
Rekonstituce s použitím BAXJECT II
Rekonstituce s použitím jehel
1. Zahřejte Recombinate (prášek) a sterilizovanou vodu pro injekci na teplotu 15–25°C
1. Zahřejte Recombinate (prášek) a sterilizovanou vodu pro injekci na teplotu 15–25°C
2. Sejměte uzávěry z lahviček s práškem a rozpouštědlem.
2. Sejměte uzávěry z lahviček s práškem a rozpouštědlem.
3. Zátky očistěte dezinfekčními polštářky. Lahvičky umístěte na rovný povrch.
3. Zátky očistěte dezinfekčními polštářky. Lahvičky umístěte na rovný povrch.
4. Odtržením papírového krytu otevřete obal pomůcky BAXJECT II, aniž byste se dotkl(a) vnitřku (Obr. A) Pomůcku nevyjímejte z obalu.
4. Sejměte ochranný kryt z jednoho konce přepouštěcí jehly se dvěma konci a propíchněte jím zátku lahvičky s rozpouštědlem.
5. Obal převraťte a průhledným plastovým
5. Sejměte ochranný kryt z druhého konce
hrotem propíchněte zátku lahvičky s rozpouštědlem. Obal uchopte na konci a sejměte jej z pomůcky BAXJECT II (Obr. B). Nesnímejte modrý kryt pomůcky BAXJECT II.
přepouštěcí jehly se dvěma konci. Obraťte lahvičku s rozpouštědlem dnem vzhůru nad lahvičku s přípravkem Recombinate, potom rychle propíchněte volným koncem jehly střed zátky lahvičky s přípravkem Recombinate. Rozpouštědlo bude vtaženo vakuem v lahvičce.
6. S pomůckou BAXJECT II připojenou k lahvičce s rozpouštědlem převraťte celý systém tak, aby lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku přípravku Recombinate. Rozpouštědlo bude vtaženo vakuem do lahvičky s přípravkem Recombinate (Obr. C).
6. Odpojte obě lahvičky vytažením jehly z lahvičky s přípravkem Recombinate. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je Recombinate zcela rozpuštěn, jinak se aktivní materiál zachytí ve filtrační jehle.
7. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je Recombinate zcela rozpuštěn, jinak by aktivní materiál neprošel filtrem pomůcky. Přípravek se rozpouští rychle (obvykle do 1 minuty)
Obr. A Obr. B Obr. C
** V 1 r-n
podání nebo dočasném přerušení injekce. (viz body 4.4 a 4.8)
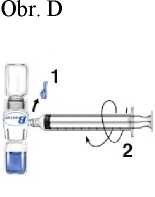
Obr. E zrychlení zpravidla rychle vymizí po zpomalení rychlosti podání nebo dočasném přerušení injekce. (viz body 4.4 a 4.8)

-
5. K natažení každé lahvičky přípravku Recombinate má být použita samostatná nepoužitá filtrační jehla.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxalta Innovations GmbH
Industriestrasse 67
A-1221 Vídeň
Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
Recombinate 500 International Unit/5 ml prášek a rozpouštědlo pro injekční roztok: 75/216/13-C
Recombinate 1000 International Unit/5 ml prášek a rozpouštědlo pro injekční roztok: 75/217/13-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
2. 5. 2013/12. 8. 2017
Další informace o léčivu RECOMBINATE 1000 INTERNATIONAL UNIT/5 ML
Jak
se RECOMBINATE 1000 INTERNATIONAL UNIT/5 ML
podává: intravenózní podání - prášek a rozpouštědlo pro injekční roztok
Výdej
léku: na lékařský předpis
Balení: Injekční lahvička
Velikost
balení: 1+1X5ML
Držitel rozhodnutí o registraci daného léku v České republice:
Baxalta Innovations GmbH, Vídeň
E-mail: objednavky.cz@shire.com
Telefon: +420 225 379 700