Souhrnné informace o léku - HAEMOCTIN SDH 1000
1. NÁZEV PŘÍPRAVKU
2.
1. NÁZEV PŘÍPRAVKU
Haemoctin SDH 250
Haemoctin SDH 500 Haemoctin SDH 1000
Prášek a rozpouštědlo pro injekční roztok
2.
Factor VIII coagulationis humanus
Jedna injekční lahvička obsahuje factor VIII coagulationis humanus 250, 500 nebo 1000 IU derivovaný z lidské plazmy.
Po rekonstituci se 5 ml nebo 10 ml vody na injekce obsahuje Haemoctin SDH 250 nebo Haemoctin SDH 500 přibližně 50 IU/ml lidského koagulačního faktoru VIII. Po rekonstituci s 10 ml vody na injekce obsahuje Haemoctin SDH 1000 přibližně 100 IU/ml lidského koagulačního faktoru VIII.
Síla (IU) se určuje pomocí chromogenního testu k určení koagulačního faktoru VIII podle Evropského lékopisu. Specifická aktivita přípravku Haemoctin SDH je přibližně 100 IU/mg proteinu.
Vyrobeno z plazmy lidských dárců.
Pomocná látka se známým účinkem.
Jedna injekční lahvička obsahuje až 32,2 mg sodíku (1,4 mmol).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Bílý prášek a čiré, bezbarvé rozpouštědlo pro injekční roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (dědičný nedostatek faktoru VIII).
Přípravek neobsahuje von Willebrandův faktor ve farmakologicky účinném množství a není tedy indikován na von Willebrandovu nemoc.
4.2 Dávkování a způsob podání
Léčba má být pod dohledem lékaře, který má zkušenosti s léčbou hemofilie.
Dosud neléčení pacienti
Nejsou dostupné žádné údaje.
Sledování léčby
Během léčby se doporučuje stanovit hladiny faktoru VIII, aby bylo známo, jakou dávku podávat a jak často infuze opakovat. U jednotlivých pacientů se může jejich odpověď na faktor VIII lišit, což se projeví odlišnými poločasy a dobami zotavení. Dávka založená na tělesné hmotnosti může vyžadovat úpravu u pacientů s podváhou nebo nadváhou. Zejména v případě velkých chirurgických zákroků je nezbytné přesné monitorování substituční léčby pomocí koagulačních testů (aktivita plazmatického faktoru VIII).
Při použití jednostupňového testu srážlivosti na základě tromboplastinového času (aPTT) in vitro ke stanovení aktivity faktoru VIII ve vzorcích krve pacienta mohou být výsledky aktivity faktoru VIII významně ovlivněny jak typem reagencie aPTT, tak referenčním standardem použitým v testu. Rovněž může dojít k významným nesrovnalostem u výsledků získaných jednostupňovým testem srážlivosti na základě tromboplastinového času (aPTT) a chromogenním testem podle Evropského lékopisu. To je mimořádně důležité při změně laboratoře a/nebo reagencií použitých v testu.
Dávkování
Dávka a délka trvání substituční léčby závisí na závažnosti nedostatku faktoru VIII, místě a rozsahu krvácení a klinickém stavu pacienta.
Počet jednotek podaného faktoru VIII se vyjadřuje v mezinárodních jednotkách (IU), které se vztahují k stávající koncentrované normě WHO pro přípravky na bázi faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď jako procento (ve vztahu k normální lidské plazmě), nebo nejlépe v mezinárodních jednotkách (ve vztahu k mezinárodnímu standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII je ekvivalentní množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba podle potřeby
Výpočet požadované dávky faktoru VIII je založen na empirické zkušenosti, že podání 1 mezinárodní jednotky (IU) faktoru VIII na kg tělesné hmotnosti zvýší hladinu plazmatického faktoru VIII o 1 % až 2 % normální aktivity.
Potřebnou dávku lze vypočítat podle následujícího vzorce:
Podané množství a frekvence podávání se musí řídit klinickou účinností v jednotlivých případech.
V případě následujících krvácivých událostí nesmí aktivita faktoru VIII klesnout pod danou hladinu aktivity plazmy (v % normální hodnoty) po odpovídající dobu. Následující tabulku lze použít jako návod dávkování u krvácivých epizod a při chirurgických výkonech:
= . v s ---
Stupeň krvácení / Požadovaná hladina Četnost dávek (v hodinách) /
typ chirurgického výkonu faktoru VIII (%) trvání léčby (ve dnech)
Krvácení
Počáteční hemartróza, 20 – 40
krvácení do svalů nebo
orální krvácení
Extenzivnější hemartróza, 30 – 60
krvácení do svalů nebo
hematom
Opakovat každých 12 až 24 hodin. Nejméně 1 den, až do vyřešení krvácivé epizody indikované bolestí nebo do zhojení příslušného zranění.
Opakovat každých 12 až 24 hodin po dobu 3–4 dnů, nebo až do
vyřešení bolestivého a akutního stavu.
Život ohrožující krvácení 60 – 100 Opakovat každých 8 až 24 hodin až
stav ohrožení pomine.
| Operace | ||
| Malé operace včetně extrakce zubů | 30 – 60 | Každých 24 hodin, alespoň 1 den, až do zhojení. |
| Velké operace | 80 – 100 (před a po operaci) | Opakovat každých 8 až 24 hodin, až do adekvátního zhojení ran, |
potom léčba po dobu alespoň 7 dalších dnů k udržení aktivity faktoru VIII v hladině 30 až 60 %.
Profylaxe
Při dlouhodobé profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na kg tělesné hmotnosti v 2 až 3denních intervalech. V některých případech, zvláště u mladších pacientů, mohou být nezbytné kratší intervaly mezi aplikacemi nebo vyšší dávky.
Způsob podání
Intravenózní podání. Nedoporučuje se podávat více než 2–3 ml přípravku Haemoctin SDH za minutu. Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
Jako u každého intravenózně podávaného proteinového přípravku se mohou vyskytnout hypersenzitivní reakce typu alergií. Přípravek obsahuje stopy lidských bílkovin, které jsou odlišné od faktoru VIII. Pro případ, že by se objevily příznaky hypersenzitivity, je třeba pacienty poučit, aby přípravek okamžitě přestali používat a kontaktovali lékaře. Je třeba informovat pacienty, že se mohou vyskytnout příznaky hypersenzitivních reakcí, jako například vyrážka, generalizovaná kopřivka, svíravý pocit na hrudi, dušnost, hypotenze a anafylaxe.
V případě šoku je třeba postupovat podle stávajících standardních léčebných metod pro léčbu šoku.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle imunoglobuliny IgG zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. Riziko vzniku inhibitorů souvisí se závažností onemocnění i s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů, s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze, z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Klinický význam vzniku inhibitorů bude záviset na titru inhibitoru, přičemž inhibitory nízkého titru, které jsou krátkodobě přítomny nebo zůstávají trvale na nízkém titru, představují menší riziko nedostatečné klinické odpovědi než inhibitory vysokého titru.
Obecně platí, že všichni pacienti léčení přípravky s koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být terapie faktorem VIII účinná a je třeba zvážit jiné možnosti léčby. Péče o takové pacienty má být vedena lékaři se zkušenostmi v péči o hemofilii a inhibitory faktoru VIII.
Kardiovaskulární příhody
U pacientů s existujícími kardiovaskulárními rizikovými faktory může substituční léčba faktorem VIII zvyšovat kardiovaskulární riziko.
Komplikace související s katetrem
Jestliže je třeba použít centrální žilní vstup (central venous access devices, CVAD), musí se zvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriemie a trombózy v místě vstupu katetru.
Přenosné látky
Mezi standardní opatření zabraňující přenosu infekcí v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy patří výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a zařazení účinných výrobních kroků zaměřených na inaktivaci/odstranění virů. Ani přes tato opatření nelze možnost přenosu infekcí v případě podávání léčivých přípravků vyrobených z lidské krve nebo plazmy zcela vyloučit. To platí také pro neznámé nebo nově objevené viry a jiné patogeny.
Přijatá opatření jsou považována za účinná u obalených virů, jako je virus lidské imunitní nedostatečnosti (HIV), virus hepatitidy B (HBV) a virus hepatitidy v (HCV), a u neobaleného viru hepatitidy A (HAV). Přijatá opatření mají omezenou účinnost u neobalených virů, jako je například parvovirus B19.
Infekce parvovirem B19 může být závažná u těhotných žen (infekce plodu) a u jedinců s imunodeficitem nebo zvýšenou erytropoézou (např. hemolytická anémie).
U pacientů, kteří dostávají pravidelně/opakovaně léky na bázi faktoru VIII derivované z lidské plazmy, je třeba zvážit vhodnou vakcinaci (hepatitida A a B).
Důrazně se doporučuje, aby byl při každém podání přípravku Haemoctin SDH pacientovi zaznamenán název a číslo šarže přípravku, aby se zachovala vazba mezi pacientem a použitou šarží přípravku.
Pediatrická populace
Zvláštní upozornění a opatření pro použití uvedené pro dospělé mají být zvažována také pro pediatrickou populaci.
Obsah sodíku
Jedna injekční lahvička obsahuje až 32,2 mg sodíku (1,4 mmol). Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nejsou známé žádné interakce lidského koagulačního faktoru VIII s dalšími léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
S faktorem VIII nebyly provedeny reprodukční studie na zvířatech. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou k dispozici zkušenosti o používání faktoru VIII během těhotenství nebo kojení. Faktor VIII tedy nemá být během těhotenství nebo kojení užíván, pokud není zřetelně indikován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Haemoctin SDH nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
V některých případech byly vzácně pozorovány hypersenzitivní nebo alergické reakce (například angioedém, pálení a píchání v místě aplikace infuze, zimnice, zčervenání, generalizovaná kopřivka, bolest hlavy, vyrážka, hypotenze, letargie, nevolnost, neklid, tachykardie, svíravý pocit na hrudi, brnění, zvracení nebo dušnost) a v některých případech vedly k závažné anafylaxi (například i šok).
K rozvoji neutralizujících protilátek (inhibitorů) může dojít u pacientů s hemofilií A, kteří jsou léčeni faktorem VIII, včetně přípravku Haemoctin. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum.
Informace o bezpečnosti s hlediska přenosných agens naleznete v bodu 4.4.
Tabulkový přehled nežádoucích účinků
Údaje v tabulce níže jsou uvedeny podle klasifikace orgánových systémů MedDRA (SOC a Preferred Term Level).
Četnosti výskytu nežádoucích účinků byly definovány následovně: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit)).
V klinických zkouškách, neintervenčních studiích, dobrovolných hlášeních a pravidelném screeningu literatury byly zjištěny následující nežádoucí účinky související s přípravkem Haemoctin SDH :
| Třídy orgánových systémů podle databáze MedDRA | Nežádoucí účinky | Četnost |
| Poruchy kůže a podkožní tkáně | Exantém, kopřivka, erytém | velmi vzácné |
| Poruchy krve a lymfatického systému | Inhibice faktoru VIII | Méně časté (PTP)* Velmi časté (PUP)* |
Četnost vychází ze studií se všemi přípravky s faktorem VIII, které zahrnovaly pacienty se závažnou hemofilií A. PTP = dříve léčení pacienti, PUP = dříve neléčení pacienti
Pediatrická populace
Četnost, typ a závažnost nežádoucích účinků u pediatrické populace mají být stejné jako u dospělých.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
Nebyl hlášen žádný případ předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika: krevní koagulační faktor VIII ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktor VIII a von Willebrandův faktor) s odlišnými fyziologickými funkcemi.
Po aplikaci infuze pacientovi s hemofilií se faktor VIII naváže na von Willebrandův faktor v pacientově krevním oběhu.
Aktivovaný faktor VIII se chová jako kofaktor pro aktivovaný faktor IX, který zrychluje konverzi faktoru X na aktivovaný faktor X (faktor Xa).
Aktivovaný faktor X pak konvertuje protrombin na trombin. Trombin konvertuje fibrinogen na fibrin a ten vytváří krevní sraženinu. Hemofilie A je na pohlaví závislá dědičná porucha srážení krve způsobená sníženými hladinami faktoru VIII:C a má za následek profúzní krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánně, nebo jako následek operačního traumatu. Substituční léčbou se zvýší hladiny faktoru VIII v plazmě, tím se dočasně umožní korigovat nedostatek faktoru a sklon ke krvácení.
Von Willebrandův faktor, jako ochranný protein faktoru VIII, zprostředkovává adhezi krevních destiček na cévní endotel a hraje důležitou roli v agregaci krevních destiček.
U pacientů s hemofilií A, u nichž se vyvinuly inhibitory faktoru VIII, byla úspěšně shromážděna data o provedené indukci tolerance imunity (ITI).
5.2 Farmakokinetické vlastnosti
Plazmatická aktivita faktoru VIII po intravenózním podání klesá v dvoufázové exponenciální křivce. Během první fáze probíhá distribuce mezi intravaskulárními a dalšími kompartmenty (tělesnými tekutinami) s poločasem eliminace z plazmy 1 až 8 hodin. V následující pomalejší fázi kolísá poločas mezi 5–18 hodinami s průměrem přibližně 12 hodin. Zdá se, že tento údaj odpovídá skutečnému biologickému poločasu.
Inkrementální výtěžnost přípravku Haemoctin SDH je přibližně 0,020 ± 0,003 IU/ml/IU/kg těl. hmotnosti. Hladina aktivity faktoru VIII po intravenózním podání 1 IU faktoru VIII na kg tělesné hmotnosti je přibližně 2 %.
Další farmakokinetické parametry přípravku Haemoctin SDH jsou:
- Oblast pod křivkou (AUC): asi 17 IU x hod/ml
- Střední rezidenční čas (mean residence time, MRT): přibližně 15 hod
- Clearance: přibližně 155 ml/hod.
5.3 Předklinické údaje vztahující se k bezpečnosti
Lidský plazmatický koagulační faktor VIII (z koncentrátu) obsažený v přípravku je normální složkou lidské plazmy a účinkuje stejně jako endogenní faktor VIII. Testování toxicity jedné dávky není relevantní vzhledem k přetížení organismu. Testování toxicity opakovanou dávkou u zvířat je neproveditelné vzhledem k indukci tvorby protilátek k cizorodému proteinu.
Dávky několikrát převyšující v přepočtu na kilogram tělesné hmotnosti doporučené dávky u lidí nevyvolávají u zvířat žádné toxické účinky.
Protože klinické zkušenosti nenaznačují žádný tumorogenní nebo mutagenní efekt lidského plazmatického koagulačního faktoru VIII, nejsou experimentální studie, zvláště na heterologních druzích, považovány za nutné.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek: Glycin, chlorid sodný, natrium-citrát, chlorid vápenatý Rozpouštědlo: Voda na injekce.
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
K aplikaci smí být použity pouze přiložené infuzní sety, protože vlivem adsorpce faktoru VIII k vnitřnímu povrchu některých součástí infuzního setu by mohlo dojít k selhání léčby.
6.3 Doba použitelnosti
2 roky
Po prvním otevření se přípravek musí použít okamžitě.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
Injekční lahvičky uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Obsah 1 balení přípravku Haemoctin SDH:
1 injekční lahvička s práškem (20 ml), sklo typu I dle Ph. Eur.
Lyofilizované zátky z halobutylové pryže, typ I dle Ph. Eur.
1 injekční lahvička s rozpouštědlem (5 ml, 10 ml), sklo typu I dle Ph. Eur.
Zátky injekce z halobutylové pryže, typ I dle Ph. Eur.
Balení obsahuje také:
1 jednorázovou injekční stříkačku (5 ml, 10 ml), 1 převodní zařízení s filtrem, 1 infuzní kanylu (motýlek).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Rekonstituovaný léčivý přípravek musí být před podáním vizuálně zkontrolován pro výskyt částic a změnu barvy. Roztok musí být čirý nebo lehce opalescentní. Nepoužívejte roztoky, které jsou zakalené nebo obsahují usazeniny.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky
Návod k použití přípravku a zacházení s ním:
Je třeba po celou dobu postupu zachovávat absolutní sterilitu!
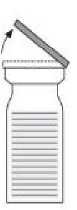
Fig. 1
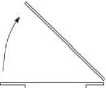
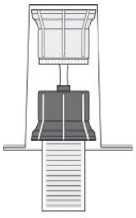
Fig. 3
Fig. 2
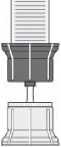

Fig. 4
- Ohřejte rozpouštědlo (vodu na injekci)
a prášek na pokojovou teplotu v neotevřených injekčních lahvičkách. Pokud je používána k zahřívání vodní lázeň, je nutno úzkostlivě dbát, aby voda nepřišla do kontaktu s víčkem nebo zátkou na injekční lahvičky. Jinak může dojít ke kontaminaci přípravku.
- Odstraňte víčka z obou injekčních lahviček tak, aby se odkryla centrální část pryžové zátky (1). Očistěte zátky přípravku a injekčních lahviček
s rozpouštědlem dezinfekčním prostředkem.
- Sejměte uzávěr obalu převodního zařízení (2). Umístěte modrou část převodního zařízení na svisle stojící injekční lahvičku obsahující rozpouštědlo (3).
- Odstraňte zbývající část obalu převodního zařízení. Tak se odhalí průhledná část převodního zařízení.
- Položte injekční lahvičku s přípravkem na rovný povrch.

Fig. 5
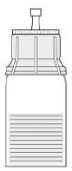
Fig. 6
- Obraťte převodní zařízení s injekční lahvičkou s rozpouštědlem dnem vzhůru. Vtlačte hrot průhledné části adaptéru kolmo dolů skrz zátku injekční lahvičky přípravku (4). Vakuum přítomné
v injekční lahvičce s přípravkem způsobí, že rozpouštědlo proteče do injekční lahvičky s přípravkem (5). Okamžitě vyšroubujte modrou část převodního zařízení zároveň s injekční lahvičkou s rozpouštědlem. Zlikvidujte injekční lahvičku s rozpouštědlem s modrou částí připojeného převodního zařízení (6). Opatrné otáčení injekční lahvičky napomůže v rozpouštění prášku.
S lahvičkou netřeste, je nutné se vyhnout tvorbě pěny! Roztok je čirý nebo lehce opalescentní.
- Rozpuštěný roztok se musí použít ihned po rozpuštění. Roztoky se zákalem nebo viditelnými částicemi se nesmí použít.
- Po rozpuštění prášku podle popisu výše, našroubujte přiloženou injekční stříkačku konektorem typu luer-lock na injekční lahvičku s průhlednou částí převodního zařízení (7). Pak je možné natáhnout rozpuštěný přípravek snadno do injekční stříkačky. Není potřeba zvláštní filtr, neboť převodní zařízení má zabudovaný vlastní filtr.
- Opatrně odpojte injekční lahvičku s průhlednou částí převodního zařízení od injekční stříkačky. Přípravek pomalu intravenózně aplikujte pomocí přiložené motýlkové jehly. Rychlost podání injekce nesmí přesáhnout 2–3 ml za minutu.
- Po použití motýlkové kanyly použijte ochranné víčko, aby byla další manipulace bezpečná.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Biotest Pharma GmbH
Landsteinerstrasse 5
63303 Dreieich
Německo
Tel.: +49 6103 801–0
Fax: +49 6103 801–150
E-mail:
8. REGISTRAČNÍ ČÍSLO(A)
Haemoctin SDH 250: 75/139/92-A/C
Haemoctin SDH 500: 75/139/92-B/C
Haemoctin SDH 1000: 75/139/92-C/C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 5. 2. 1992
Datum posledního prodloužení registrace: 16. 10. 2012
Další informace o léčivu HAEMOCTIN SDH 1000
Jak
se HAEMOCTIN SDH 1000
podává: intravenózní podání - prášek a rozpouštědlo pro injekční roztok
Výdej
léku: na lékařský předpis
Balení: Injekční lahvička
Velikost
balení: 1+1X10ML
Držitel rozhodnutí o registraci daného léku v České republice:
Biotest Pharma GmbH, Dreieich
E-mail: infoservis@regpharm.cz
Telefon: 272654004