Souhrnné informace o léku - DAYLETTE 3 MG/0,02 MG POTAHOVANÉ TABLETY
1.
Daylette 3 mg/0,02 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
24 bílých až téměř bílých (aktivních) potahovaných tablet:
Jedna potahovaná tableta obsahuje drospirenonum 3 mg a ethinylestradiolum 0,02 mg.
Pomocné látky se známým účinkem: jedna potahovaná tableta obsahuje monohydrát laktosy 48,53 mg a sójový lecithin 0,070 mg.
4 zelené placebo (neaktivní) potahované tablety:
Tablety neobsahují léčivé látky.
Pomocné látky se známým účinkem: laktosa 37,26 mg, hlinitý lak oranžové žluti (E110) 0,003 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Aktivní tableta je bílá až téměř bílá, kulatá, bikonvexní potahovaná tableta, průměr 6 mm. Na jedné straně vyraženo “G73”, na druhé straně hladká.
Placebo tableta je zelená, kulatá, bikonvexní potahovaná tableta, průměr 6 mm, obě strany hladké.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Perorální kontracepce.
Rozhodnutí předepsat přípravek Daylette by mělo být provedeno po zvážení jednotlivých současných rizikových faktorů ženy, zvláště rizikových faktorů pro žilní tromboembolismus (VTE), a toho, jaké je riziko VTE u přípravku Daylette v porovnání s dalšími přípravky kombinované hormonální kontracepce (CHC) (viz body 4.3 a 4.4).
4.2 Dávkování a způsob podání
Způsob podání: perorální podání
Dávkování
Tablety se musí užívat každý den přibližně ve stejnou dobu v pořadí vyznačeném na blistru, podle potřeby se zapíjejí tekutinou. Užívání tablet je kontinuální. Užívá se jedna tableta denně po dobu 28 po sobě následujících dnů. Užívání z nového blistru se začíná další den po užití poslední tablety z předchozího blistru. Krvácení z vysazení začíná obvykle 2. – 3. den užívání zelených placebo tablet (poslední řada) a nemusí být ukončeno v době zahájení užívání z dalšího blistru.
- Nepředcházelo-li užívání hormonální kontracepce (v předchozím měsíci)
- Přechod z kombinovaného hormonálního kontraceptiva (kombinované perorální kontraceptivum (COC), vaginální kroužek nebo transdermální náplast)
- Přechod z kontracepční metody obsahující pouze progestogen (pilulka s progestogenem, injekce, implantát) nebo z nitroděložního systému uvolňujícího progestogen (IUS)
- Užívání po potratu v prvním trimestru
- Užívání po porodu nebo po potratu ve druhém trimestru
Vynechání placebových tablet (zelených tablet) z poslední (4.) řádky blistru není třeba věnovat zvláštní pozornost. Je však třeba je zlikvidovat, aby nedošlo k neúmyslnému prodloužení placebové fáze. Následující pokyn platí pouze pro vynechané aktivní tablety:
Pokud se užití tablety opozdí o méně než 24 hodin, není kontracepční ochrana snížena. Žena musí užít tabletu okamžitě, jakmile si vzpomene a další tablety pak užije v obvyklou dobu.
Je-li užití tablety opožděno o více než 24 hodin, kontracepční ochrana může být snížena. Další opatření se pak mohou řídit následujícími dvěma základními pravidly:
-
1. Doporučená délka intervalu užívání placebo tablet je 4 dny, užívání tablet nesmí být nikdy přerušeno na dobu delší než 7 dní.
-
2. K dosažení odpovídající suprese hypothalamo-hypofyzo-ovariální osy je třeba 7 dnů nepřerušeného užívání tablet.
-
V souladu s těmito pravidly lze v běžné praxi poskytnout následující doporučení:
Uživatelka musí užít poslední vynechanou tabletu okamžitě, jakmile si vzpomene, i kdyby to znamenalo užití dvou tablet současně. Poté pokračuje v užívání tablet v obvyklou dobu. Navíc je třeba používat v následujících 7 dnech bariérovou metodu kontracepce jako např. kondom. Pokud došlo v předchozích 7 dnech k pohlavnímu styku, je třeba zvážit možnost otěhotnění. Čím více tablet bylo vynecháno a čím blíže byly tyto tablety k placebové fázi, tím větší je riziko otěhotnění.
8. – 14. denUživatelka musí užít poslední vynechanou tabletu okamžitě, jakmile si vzpomene, i kdyby to znamenalo užití dvou tablet současně. Poté pokračuje v užívání tablet v obvyklou dobu. Pokud žena užívala tablety pravidelně po dobu 7 dnů před první vynechanou tabletou, další kontracepční opatření nejsou nutná. Vynechala-li však žena více než 1 tabletu, je třeba doporučit zvláštní kontracepční opatření po dobu 7 dnů.
15. – 24. denVzhledem k nadcházející placebové fázi je velké nebezpečí snížení spolehlivosti kontracepce. Přesto však upravením schématu užívání lze předejít snížení kontracepční ochrany. Bude-li se uživatelka řídit některým z následujících dvou možných postupů, není třeba používat další kontracepční opatření za předpokladu, že po dobu 7 dnů předcházejících vynechání první tablety užila všechny tablety správně. Není-li tomu tak, žena musí zvolit první z následujících dvou možností a použít navíc další kontracepční opatření po dobu 7 dnů.
-
1. Uživatelka musí užít poslední vynechanou tabletu okamžitě, jakmile si vzpomene, i kdyby to znamenalo užití dvou tablet současně. Poté pokračuje v užívání tablet v obvyklou dobu, dokud nevyužívá aktivní tablety. 4 zelené placebové tablety z poslední řádky je nutno vyřadit. Užívání z následujícího blistru pak zahájí okamžitě po využívání předchozího. Krvácení z vysazení se pravděpodobně dostaví až po využívání aktivních tablet z druhého blistru, ale během užívání tablet může dojít ke špinění nebo krvácení z průniku.
-
2. Ženě lze také poradit, aby přerušila užívání aktivních tablet ze stávajícího blistru. Poté by měla užívat zelené placebo tablety z poslední řádky po dobu 4 dní – včetně dnů, kdy byly tablety vynechány, a poté začne užívat další blistr.
Pokud žena zapomene užít tablety a následně se nedostaví krvácení z vysazení v době užívání placebo tablet, je třeba zvážit možnost těhotenství.
-
V případě závažnějších gastrointestinálních poruch (například zvracení nebo průjmu) nemusí dojít k úplnému vstřebání a je třeba použít další kontracepční opatření.
Dojde-li do 3 – 4 hodin po užití aktivní tablety ke zvracení, je třeba co nejdříve užít novou (náhradní) tabletu. Nová tableta se má, pokud možno, užít do 24 hodin od obvyklé doby užívání. Pokud uplynulo více než 24 hodin, lze aplikovat postup při vynechání tablety uvedený v bodě 4.2 „Postup při vynechání tablet“. Nechce-li žena měnit obvyklé schéma užívání tablet, musí užít zvláštní tabletu (y) z jiného blistru.
Přeje-li si žena oddálit krvácení, musí pokračovat v užívání tablet z dalšího blistru přípravku Daylette bez užívání placebo tablet ze současného blistru. Tak lze pokračovat podle přání až do využívání
3/18
aktivních tablet z druhého blistru. Během této doby může žena pozorovat krvácení z průniku nebo špinění. Po užití placebo tablet pak žena opět pokračuje v pravidelném užívání přípravku
Daylette.
Přeje-li si žena přesunout periodu na jiný den v týdnu, než na který vychází ve stávajícím schématu užívání, lze jí doporučit, aby zkrátila nastávající fázi placebo tablet o tolik dnů, o kolik si přeje. Čím kratší bude interval, tím větší je riziko, že nedojde ke krvácení z vysazení, ale že bude docházet během užívání z následujícího blistru ke krvácení z průniku a špinění (podobně jako při oddálení periody).
4.3 Kontraindikace
Kombinovaná hormonální kontraceptiva (CHC) nelze užívat, je-li u pacientky diagnostikován některý z dále uvedených stavů. Pokud se některý z těchto stavů objeví poprvé v průběhu užívání CHC, užívání přípravku je třeba okamžitě ukončit.
-
– Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
-
– Hypersenzitivita na burské oříšky nebo sóju.
-
– Přítomnost nebo riziko žilního tromboembolismu (VTE)
-
– žilní tromboembolismus – současný žilní tromboembolismus (léčený pomocí antikoagulancií) nebo anamnéza VTE (např. hluboká žilní trombóza [DVT] nebo plicní embolie [PE])
-
– známá dědičná nebo získaná predispozice pro žilní tromboembolismus, jako je rezistence na APC (včetně faktoru V Leiden), deficit antitrombinu III, deficit proteinu C, deficit proteinu S
-
– velký chirurgický zákrok s déletrvající imobilizací (viz bod 4.4)
-
– vysoké riziko žilního tromboembolismu v důsledku přítomnosti více rizikových faktorů (viz bod 4.4);
-
– Přítomnost nebo riziko arteriálního tromboembolismu (ATE)
-
– arteriální tromboembolismus – současný arteriální tromboembolismus, anamnéza arteriálního tromboembolismu (např. infarkt myokardu) nebo prodromální stav (např. angina pectoris);
-
– cerebrovaskulární onemocnění – současná cévní mozková příhoda, anamnéza cévní mozkové příhody nebo prodromálního stavu (např. tranzitorní ischemická ataka, TIA);
-
– známá hereditární nebo získaná predispozice k arteriálnímu tromboembolismu, jako je hyperhomocysteinemie a antifosfolipidové protilátky (antikardiolipinové protilátky, lupus antikoagulans);
-
– anamnéza migrény s fokálními neurologickými příznaky;
-
– vysoké riziko arteriálního tromboembolismu v důsledku vícečetných rizikových faktorů (viz bod 4.4) nebo přítomnosti jednoho závažného rizikového faktoru, jako je:
-
– diabetes mellitus s cévními příznaky;
-
– závažná hypertenze;
-
– závažná dyslipoproteinemie.
-
– Těžké jaterní onemocnění právě probíhající nebo v anamnéze až do navrácení hodnot jaterních funkcí k normálu.
-
– Závažná renální insuficience nebo akutní renální selhání.
-
– Existující jaterní tumory nebo jejich výskyt v anamnéze (benigní či maligní).
-
– Přítomnost pohlavními steroidy ovlivnitelných malignit (např. pohlavních orgánů nebo prsů) nebo podezření na ně.
-
– Vaginální krvácení s nediagnostikovanou příčinou.
4.4 Zvláštní upozornění a opatření pro použití
Pokud jsou přítomna jakákoli onemocnění nebo rizikové faktory uvedené níže, měla by být vhodnost přípravku Daylette s ženou prodiskutována.
-
V případě zhoršení nebo prvního výskytu jakéhokoli z těchto stavů nebo rizikových faktorů by mělo být ženě doporučeno, aby kontaktovala svého lékaře, který stanoví, zda by měla užívání přípravku Daylette ukončit.
-
V případě podezření nebo potvrzení žilního tromboembolismu (VTE) nebo arteriálního tromboembolismu (ATE) musí být přerušeno užívání kombinované hormonální antikoncepce (CHC).
-
V případě, že je antikoagulační léčba zahájena, měla by být užívána adekvátní alternativní antikoncepce a to z důvodu teratogenních účinků antikoagulační léčby (kumariny).
Riziko žilního tromboembolismu (VTE)
Užívání jakékoli kombinované hormonální antikoncepce (CHC) zvyšuje riziko žilního tromboembolismu (VTE) ve srovnání s jejím neužíváním. Přípravky, které obsahují levonorgestrel, norgestimát nebo norethisteron jsou spojovány s nejnižším rizikem VTE. Další přípravky, jako je přípravek Daylette mohou mít až dvakrát vyšší úroveň rizika. Rozhodnutí používat jakýkoli přípravek jiný než ten, který má nejnižší riziko VTE, by mělo být učiněno po diskusi se ženou, aby se zajistilo, že rozumí riziku VTE u přípravku Daylette, rozumí, jak její současné rizikové faktory toto riziko ovlivňují a že riziko VTE je nejvyšší v prvním roce užívání léku. Existují také některé důkazy, že riziko je zvýšené, když je CHC opětovně zahájena po pauze v užívání trvající 4 týdny nebo déle.
U žen, které neužívají CHC a nejsou těhotné, se asi u 2 z 10 000 vyvine VTE v průběhu jednoho roku. U každé jednotlivé ženy však může být riziko daleko vyšší v závislosti na jejích základních rizikových faktorech (viz níže).
Odhaduje se1 že z 10 000 žen, které používají CHC obsahující drospirenon se u 9 až 12 žen vyvine VTE během jednoho roku; v porovnání s přibližně 62 případy u žen, které používají CHC obsahující levonorgestrel.
VTE může být fatální v 1–2 % případů.
Počet příhod VTE na 10 000 žen za rok
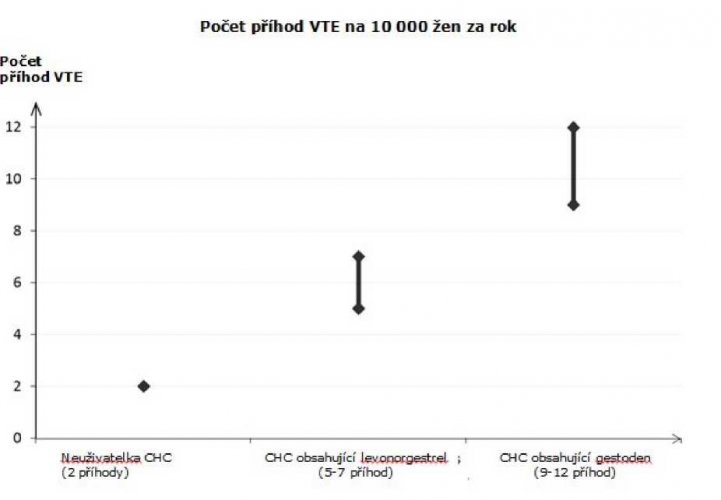
1 Tyto incidence byly odhadnuty ze souhrnu dat z epidemiologických studií s použitím relativních rizik pro různé přípravky ve srovnání s CHC obsahující levonorgestrel.
2 Střední bod rozmezí 5–7 na 10 000 WY (žen-roků) na základě relativního rizika pro CHC obsahující levonorgestrel oproti jejímu nepoužívání přibližně 2,3 až 3,6
Extrémně vzácně byla hlášena trombóza u uživatelek CHC v dalších cévách, např. jaterních, mezenterických, renálních nebo retinálních žilách a tepnách.
Riziko žilních tromboembolických komplikací u uživatelek CHC se může podstatně zvyšovat u ženy, která má další rizikové faktory, zvláště pokud je přítomno více rizikových faktorů (viz tabulka).
Přípravek Daylette je kontraindikován, pokud má žena více rizikových faktorů, které pro ni představují vysoké riziko žilní trombózy (viz bod 4.3). Pokud má žena více než jeden rizikový faktor, je možné, že zvýšení rizika je vyšší než součet jednotlivých faktorů – v tomto případě by mělo být zváženo její celkové riziko VTE. Pokud je poměr přínosů a rizik považován za negativní, neměla by být CHC předepisována (viz bod 4.3).
Tabulka: Rizikové faktory VTE
| Rizikový faktor | Poznámka |
| Obezita (index tělesné hmotnosti nad 30 kg/m2) | Při zvýšení BMI se významně zvyšuje riziko. Zvláště důležité je zvážit, zda jsou také přítomny další rizikové faktory. |
| Prodloužená imobilizace, velký chirurgický zákrok, jakýkoli chirurgický zákrok na nohách a pánvi, neurochirurgický zákrok nebo větší | V těchto situacích je doporučeno ukončit používání/užívání náplasti/antikoncepčních tablet/kroužku (v případě plánovaného chirurgického výkonu minimálně 4 týdny předem) a nezahajovat |
| trauma. Poznámka: dočasná imobilizace, včetně cestování letadlem > 4 hodiny může být také rizikovým faktorem VTE, zvláště u žen s dalšími rizikovými faktory | užívání do dvou týdnů po kompletní remobilizaci. Měla by se použít další antikoncepční metoda pro zabránění nechtěnému těhotenství. Antitrombotická léčba by měla být zvážena, pokud přípravek Daylette nebyl předem vysazen. |
| Pozitivní rodinná anamnéza (žilní tromboembolismus kdykoli u sourozence nebo rodiče, zvláště v relativně nízkém věku např. do 50 let věku). | Pokud je suspektní hereditární predispozice, měla by být žena před rozhodnutím o používání jakékoli CHC odeslána k odborníkovi na konzultaci |
| Další onemocnění související s VTE | Zhoubné onemocnění, systémový lupus erythematodes, hemolyticko-uremický syndrom, chronické zánětlivé onemocnění střev (Crohnova choroba nebo ulcerózní kolitida) a srpkovitá anemie |
| Vyšší věk | Zvláště nad 35 let |
Není žádná shoda o možné roli varixů a povrchové tromboflebitidy v nástupu nebo progresi žilní trombózy.
Zvýšené riziko tromboembolismu v těhotenství a zvláště během šestinedělí musí být zváženo (pro informaci o „Fertilita, těhotenství a kojení “ viz bod 4.6).
V případě příznaků by mělo být ženě doporučeno, aby vyhledala naléhavou lékařskou péči a informovala lékaře, že užívá CHC.
Příznaky hluboké žilní trombózy (DVT) mohou zahrnovat:
-
– jednostranný otok nohy a/nebo chodidla nebo podél žíly v noze;
-
– bolest nebo citlivost v noze, která může být pociťována pouze vstoje nebo při chůzi;
-
– zvýšenou teplotu postižené nohy, zarudnutí nebo změnu barvy kůže nohy.
-
– náhlý nástup nevysvětlitelné dušnosti nebo rychlého dýchání;
-
– náhlý kašel, který může být spojený s hemoptýzou;
-
– ostrou bolest na hrudi;
-
– těžké točení hlavy nebo závrať způsobené světlem;
-
– rychlý nebo nepravidelný srdeční tep.
Některé z těchto příznaků (např. „dušnost“, „kašel“) nejsou specifické a mohou být nesprávně interpretovány jako častější nebo méně závažné příhody (např. infekce dýchacího traktu).
Dalšími známkami cévní okluze mohou být: náhlá bolest, otok a světle modré zbarvení končetin. Pokud nastane okluze v oku, mohou se příznaky pohybovat od nebolestivého rozmazaného vidění, které může přejít do ztráty zraku. Někdy může nastat ztráta zraku téměř okamžitě.
Epidemiologické studie spojovaly používání CHC se zvýšeným rizikem arteriálního tromboembolismu (infarkt myokardu) nebo cerebrovaskulární příhody (např. tranzitorní ischemická ataka, cévní mozková příhoda). Arteriální tromboembolické příhody mohou být fatální.
Rizikové faktory ATE
Riziko arteriálních tromboembolických komplikací nebo cerebrovaskulární příhody u uživatelek CHC se zvyšuje u žen s rizikovými faktory (viz tabulka). Přípravek Daylette je kontraindikován, pokud má žena jeden závažný rizikový faktor nebo více rizikových faktorů ATE, které pro ni představují riziko arteriální trombózy (viz bod 4.3). Pokud má žena více než jeden rizikový faktor, je možné, že zvýšení rizika je vyšší než součet jednotlivých faktorů – v tomto případě by mělo být zváženo její celkové riziko. Pokud je poměr přínosů a rizik považován za negativní, neměla by být CHC předepisována (viz bod 4.3).
Tabulka: Rizikové faktory ATE
| Rizikový faktor | Poznámka |
| Vyšší věk | Zvláště nad 35 let |
| Kouření | Ženě by mělo být doporučeno, aby nekouřila, pokud chce používat CHC. Ženám ve věku nad 35 let, které dále kouří, by mělo být důrazně doporučeno, aby používaly jinou metodu antikoncepce. |
| Hypertenze | |
| Obezita (index tělesné hmotnosti nad 30 kg/m2) | Při zvýšení BMI se významně zvyšuje riziko. Zvláště důležité u žen s dalšími rizikovými faktory |
| Pozitivní rodinná anamnéza (arteriální tromboembolismus kdykoli u sourozence nebo rodiče, zvláště v relativně nízkém věku např. do 50 let věku). | Pokud je suspektní hereditární predispozice, měla by být žena odeslána k odborníkovi na konzultaci před rozhodnutím o používání jakékoli CHC |
| Migréna | Zvýšení frekvence nebo závažnosti migrény během používání CHC (což může být prodromální známka cévní mozkové příhody) může být důvodem okamžitého ukončení léčby |
| Další onemocnění související s nežádoucími cévními příhodami | Diabetes mellitus, hyperhomocysteinemie, chlopenní srdeční vada a fibrilace síní, dyslipoproteinemie a systémový lupus erythematodes. |
V případě příznaků by mělo být ženě doporučeno, aby vyhledala naléhavou lékařskou péči a informovala lékaře, že užívá CHC.
Příznaky cévní mozkové příhody mohou zahrnovat:
-
– náhlou necitlivost nebo slabost obličeje, paže nebo nohy, zvláště na jedné straně těla;
-
– náhlé potíže s chůzí, závratě, ztrátu rovnováhy nebo koordinace;
-
– náhlou zmatenost, problémy s řečí nebo porozuměním;
-
– náhlé potíže se zrakem na jednom nebo obou očích;
-
– náhlou, závažnou nebo prodlouženou bolest hlavy neznámé příčiny;
-
– ztrátu vědomí nebo omdlení s nebo bez záchvatu.
-
– bolest, nepříjemný pocit, tlak, těžkost, pocit stlačení nebo plnosti na hrudi, v paži nebo pod hrudní kostí;
-
– nepříjemný pocit vyzařující do zad, čelisti, hrdla, paže, žaludku;
-
– pocit plnosti, poruchu trávení nebo dušení;
-
– pocení, nauzeu, zvracení nebo závratě;
-
– extrémní slabost, úzkost nebo dušnost;
-
– rychlý nebo nepravidelný srdeční tep.
- Nádory
- Ostatní stavy
Před prvním užíváním nebo znovuzahájením užívání přípravku Daylette by měla být získána kompletní anamnéza (včetně rodinné anamnézy) a musí být vyloučeno těhotenství. Měl by se změřit krevní tlak a mělo by být provedeno tělesné vyšetření při zvážení kontraindikací (viz bod 4.3) a varování (viz bod 4.4). Je důležité, aby byla žena upozorněna na informace o žilní a arteriální trombóze, včetně rizika přípravku Daylette v porovnání s dalšími typy CHC, na příznaky VTE a ATE, známé rizikové faktory a co by měla dělat v případě suspektní trombózy.
Žena by také měla být informována, aby si pečlivě přečetla příbalovou informaci pro uživatele a aby dodržovala uvedené instrukce. Frekvence a povaha vyšetření by měly být založeny na stanovených postupech a upraveny podle individuálních potřeb ženy.
Ženy by měly být informovány, že hormonální antikoncepce nechrání před HIV infekcí (AIDS) a dalšími sexuálně přenosnými chorobami.
Účinnost COC může být snížena například při vynechání aktivních tablet (viz bod 4.2), v případě gastrointestinálních poruch během užívání aktivních tablet (viz bod 4.2) nebo při současném užívání dalších léků (viz bod 4.5)
Při užívání všech COC se může objevit nepravidelné krvácení (špinění nebo krvácení z průniku) a to především během prvních měsíců užívání. Z toho důvodu má hledání příčiny nepravidelného krvácení smysl až po adaptačním intervalu přibližně tří cyklů.
Pokud nepravidelné krvácení pokračuje nebo se objeví po období pravidelných cyklů, pak je třeba uvážit možnost nehormonální příčiny a provést odpovídající diagnostické kroky k vyloučení malignity nebo těhotenství. Mohou zahrnovat i kyretáž.
U některých žen nemusí dojít během placebo fáze ke krvácení z vysazení. Je-li COC užíváno podle pokynů popsaných v bodě 4.2, je nepravděpodobné, že je žena těhotná. Pokud však COC nebylo užíváno před prvním vynechaným krvácením dle uvedených pokynů nebo nedošlo-li ke krvácení z vysazení dvakrát, je třeba před dalším užíváním COC vyloučit těhotenství.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Poznámka: aby byly zjištěny možné interakce, měly by být prověřeny údaje o preskripci současně užívaných léčivých přípravků.
Účinky jiných léčivých přípravků na přípravek DayletteInterakce se mohou objevit s léky, které indukují mikrozomální enzymy, což může mít za následek zvýšenou clearance pohlavních hormonů a může vést ke krvácení z průniku a/nebo k selhání kontracepce.
Postup
Enzymová indukce může být pozorována již po několika dnech léčby. Maximální enzymová indukce je obvykle pozorována během několika týdnů. Po přerušení léčby může enzymová indukce přetrvávat po dobu okolo 4 týdnů.
Krátkodobá léčba
Ženy, které se léčí některým z enzymy indukujících léků, by měly přechodně používat navíc k COC bariérovou kontracepční metodu nebo použít jinou metodu kontracepce. Bariérová metoda musí být používána po celou dobu léčby souběžně podávaným lékem a dalších 28 dní po ukončení léčby.
Pokud léčba zasáhne do období ukončení užívání účinných tablet COC ze stávajícího blistru, placebo tablety musí být vyřazeny a ihned má být zahájeno užívání dalšího blistru COC.
Dlouhodobá léčba
Pokud je žena na dlouhodobé léčbě léčivou látkou, která indukuje jaterní enzymy, doporučuje se používat jinou spolehlivou nehormonální kontracepci.
V literatuře byly popsány následující interakce.
Látky zvyšující clearance COC (snižují účinnost COC enzymovou indukcí) například:
barbituráty, bosentan, karbamazepin, fenytoin, primidon, rifampicin a léky na HIV infekci ritonavir, nevirapin a efavirenz a zřejmě také felbamát, griseofulvin, oxkarbazepin, topiramát a přípravky obsahující třezalku tečkovanou (hypericum perforatum).
Látky s různým účinkem na clearance COC:
Při současném podávání společně s COC mnoho kombinací inhibitorů HIV proteázy a nenukleosidových inhibitorů reverzní transkriptázy včetně kombinací s HCV inhibitory může snižovat nebo zvyšovat plasmatickou koncentraci estrogenu nebo progestinů. Účinek těchto změn může být v některých případech klinicky významný.
Proto by měly být prostudovány informace o přípravku k souběžné léčbě HIV/HCV, aby byly identifikovány možné interakce, a příslušná doporučení. V případě jakýchkoli pochyb by ženy, které jsou na léčbě inhibitory proteázy nebo nenukleosidovými inhibitory reverzní transkriptázy, měly navíc použít bariérovou kontracepční metodu.
Hlavní metabolity drospirenonu v lidské plasmě jsou vytvářeny bez zapojení systému cytochromu P450. Je proto nepravděpodobné, že by inhibitory tohoto enzymového systému ovlivňovaly metabolismus drospirenonu.
Účinky přípravku Daylette na jiné léčivé přípravky
Perorální kontraceptiva mohou ovlivnit metabolismus některých jiných léčivých látek. Mohou jejich plasmatické a tkáňové koncentrace buď zvyšovat (např. cyklosporinu) nebo snižovat (např. lamotriginu).
Na základě in vitro inhibičních studií a studií interakcí in vivo na dobrovolnicích, které užívaly omeprazol, simvastatin a midazolam jako substrát, se ukázalo, že je interakce drospirenonu v dávce 3 mg s metabolismem jiných léčivých látek nepravděpodobná.
- Jiné formy interakce
- Laboratorní vyšetření
Užívání kontracepčních steroidů může ovlivnit výsledky některých laboratorních testů, včetně biochemických parametrů jaterních, thyreoidálních, adrenálních a renálních funkcí, plasmatických hladin proteinů (vazebných) např. kortikosteroidy vážící globulin a lipid/lipoproteinové frakce, parametrů metabolismu uhlovodanů a parametrů koagulace a fibrinolýzy. Změny však obvykle zůstávají v rozmezí normálních laboratorních hodnot. Drospirenon způsobuje svojí mírnou antimineralokortikoidní aktivitou zvýšení plasmatické aktivity reninu a plasmatického aldosteronu.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Přípravek Daylette není indikován během těhotenství.
Pokud během užívání přípravku Daylette dojde k otěhotnění, jeho další užívání musí být ihned ukončeno. Rozsáhlé epidemiologické studie však nezaznamenaly zvýšené riziko vrozených vad u dětí narozených ženám užívajícím COC před otěhotněním, ani teratogenní vliv COC neúmyslně užívaných v těhotenství.
Studie na zvířatech ukázaly nežádoucí účinek během těhotenství a kojení (viz bod 5.3). Na základě těchto údajů získaných na zvířatech nelze vyloučit nežádoucí hormonální působení léčivých látek. Všeobecné zkušenosti s užíváním COC během těhotenství však neprokázaly žádný konkrétní nežádoucí účinek u člověka.
Údaje týkající se užívání kombinace drospirenon/ethinylestradiol v těhotenství jsou příliš omezené, než aby bylo možno udělat závěry týkající se negativního vlivu kombinace drospirenon/ethinylestradiol na těhotenství, zdraví plodu nebo novorozence. Relevantní epidemiologické údaje ještě nejsou dostupné.
Zvýšené riziko VTE během poporodního období je třeba brát v úvahu při znovuzahájení užívání přípravku Daylette (viz bod 4.2 a 4.4).
Kojení
Kojení může být ovlivněno COC, která mohou snižovat množství a měnit složení mateřského mléka. Z toho důvodu se užívání COC obecně nedoporučuje, dokud matka dítě zcela neodstaví. Malá množství kontracepčních steroidů a/nebo jejich metabolitů mohou být vylučována do mléka během užívání COC. Tato množství mohou mít vliv na dítě.
Fertilita
Přípravek Daylette je indikován k prevenci otěhotnění. Informace o návratu fertility viz bod 5.1.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit nebo obsluhovat stroje nebyly provedeny. Nebyly pozorovány žádné účinky na schopnost řídit nebo obsluhovat stroje u uživatelek COC.
4.8 Nežádoucí účinky
Nejzávažnější nežádoucí účinky spojené s užíváním COC viz bod 4.4.
Během užívání kombinace drospirenon/ethinylestradiol byly hlášeny následující nežádoucí účinky: Následující tabulka uvádí nežádoucí účinky podle tříd orgánových systémů databáze MedDRA (MedDRA SOC). Frekvence jsou založené na údajích z klinických studií. K popisu reakcí a souvisejících stavů byly použity nejvhodnější termíny dle MedDRA.
| Třída orgánových systémů (verze MedDRA 18.0) | Frekvence nežádoucích účinků | |||
| Časté (> 1/100 až < 1/10) | Méně časté (> 1/1000 až < 1/100) | Vzácné (> 1/10000 až < 1/1000) | Není známo (z dostupných údajů nelze určit) | |
| Infekce a infestace | kandidóza | |||
| Poruchy krve a lymfatického systému | anemie trombocytemie | |||
| Poruchy imunitního systému | alergická reakce | hypersenzitivita | ||
| Endokrinní poruchy | endokrinní porucha | |||
| Poruchy metabolismu a výživy | zvýšená chuť k jídlu anorexie hyperkalemie hyponatremie | |||
| Psychiatrické poruchy | psychická labilita | deprese nervozita | anorgasmie insomnie | |
| Poruchy nervového systému | bolesti hlavy | závratě parestézie somnolence migréna | vertigo třes | |
| Poruchy oka | zánět spojivek suché oči další oční poruchy | |||
| Srdeční poruchy | tachykardie | |||
| Cévní poruchy | křečové žíly hypertenze | venózní tromboembolizmus (VTE), arteriální tromboembolizmus (ATE) flebitida porucha cév synkopa | ||
| Respirační, hrudní a mediastinální poruchy | epistaxe | |||
| Gastrointestinální poruchy | nauzea | bolesti břicha zvracení dyspepsie flatulence gastritida průjem | zvětšené břicho gastrointestinální poruchy pocit plnosti hiátová hernie orální kandidóza zácpa pocit sucha v ústech | |
| Poruchy jater a žlučových cest | bolesti žlučníku cholecystitida | |||
| Poruchy kůže a podkožní tkáně | akné svědění vyrážka | chloazma ekzém alopecie dermatitis acneiforme suchá kůže erythema nodosum hypertrichóza kožní poruchy kožní strie kontaktní dermatitida fotosenzitivní dermatitida kožní uzlíky | erythema multiforme | |
| Poruchy svalové a kosterní soustavy a pojivové tkáně | bolest v zádech bolest v končetinách svalové křeče | |||
| Poruchy reprodukčního systému a prsu | bolest na prsou metroragie* amenorea | vaginální kandidóza pánevní bolesti zvětšení prsů fibrocystické onemocnění prsu děložní/vaginální krvácení* výtok z genitálií návaly horka vaginitida poruchy menstruace dysmenorea hypomenorea menoragie suchost pochvy suspektní nález ve stěru Pap (Papanicolaou) snížení libida | dyspareunie postkoitální krvácení vulvovaginitida krvácení po pohlavním styku a z vysazení léku cysta v prsu hyperplazie prsu novotvary prsu cervikální polyp atrofie endometria ovariální cysty zvětšení dělohy | |
| Celkové poruchy a | astenie | malátnost |
| reakce v místě aplikace | zvýšené pocení edém (celkový edém, periferní edém, edém obličeje) | |||
| Vyšetření | přírůstek tělesné hmotnosti | úbytek tělesné hmotnosti |
* nepravidelnosti krvácení obvykle vymizí během další léčby
Popis vybraných nežádoucích účinků
U žen užívajících CHC bylo pozorováno zvýšené riziko arteriálních a žilních trombotických a tromboembolických příhod, včetně infarktu myokardu, cévní mozkové příhody, tranzitorních ischemických atak, žilní trombózy a plicní embolie a je podrobněji popsáno v bodě 4.4.
Následující závažné nežádoucí účinky, které byly hlášeny u žen užívajících COC, jsou rozvedeny v bodě 4.4 Zvláštní upozornění a opatření pro použití:
-
– Venózní tromboembolické poruchy
-
– Arteriální tromboembolické poruchy
-
– Hypertenze;
-
– Jaterní tumory;
-
– Výskyt nebo zhoršení stavů, pro které spojení s užíváním COC není jednoznačné: Crohnova choroba, ulcerózní kolitida, epilepsie, děložní myomy, porfyrie, systémový lupus erythematodes, herpes gestationis, Sydenhamova chorea, hemolyticko-uremický syndrom, cholestatická žloutenka;
-
– Chloasma;
-
– Akutní nebo chronické poruchy funkce jater si mohou vyžádat přerušení užívání COC, dokud se hodnoty ukazatelů jaterní funkce nevrátí do normálních hodnot.
-
– U žen s vrozeným angioedémem mohou exogenní estrogeny indukovat nebo exacerbovat symptomy angioedému.
U uživatelek perorálních kontraceptiv je lehce zvýšena frekvence diagnózy rakoviny prsu. Protože rakovina prsu je vzácná u žen do 40 let věku, počet případů navíc je malý ve vztahu k celkovému riziku rakoviny prsu. Kauzální vztah k COC není znám. Další informace viz body 4.3 a 4.4.
Interakce
Následkem interakcí jiných léků (enzymových induktorů) s perorální kontracepcí může být krvácení z průniku a/nebo selhání kontracepčního účinku (viz bod 4.5).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu
léčiv Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
S předávkováním kombinací drospirenon/ethinylestradiol nejsou dosud žádné zkušenosti. Na základě zkušeností s ostatními perorálními kombinovanými kontraceptivy by se v případě požití velkého množství aktivních tablet mohly vyskytnout tyto příznaky: nauzea, zvracení a u mladých dívek slabé vaginální krvácení. Neexistují žádná antidota a další léčba by měla být symptomatická.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: pohlavní hormony a modulátory pohlavního systému, progestogeny a estrogeny, fixní kombinace
ATC kód: G03AA12
Pearl index pro selhání metody: 0,41 (horní hranice 95% intervalu spolehlivosti: 0,85).
Celkový Pearl Index (selhání metody + selhání pacientky): 0,80 (horní hranice 95% intervalu spolehlivosti: 1,30).
Mechanismus účinku
Kontracepční účinek přípravku Daylette je založen na spolupůsobení různých faktorů. Nejdůležitější z nich je inhibice ovulace a změny endometria.
Během tři cykly trvající studie sledující inhibici ovulace a porovnávající podávání drospirenonu 3 mg/ ethinylestradiolu 0,020 mg ve 24 denním dávkovacím režimu oproti 21 dennímu dávkovacímu režimu, vykázal 24 denní režim větší potlačení vývoje folikulů. Po úmyslně vyvolaných chybách dávkování během třetího cyklu léčby se u více žen ve 21 denním režimu prokázala ovariální aktivita, včetně zvýšeného rizika ovulace, ve srovnání se ženami užívajícími 24 denní režim. Ovariální aktivita se vrátila na úroveň před léčbou během cyklu po ukončení léčby u 91,8 % žen, které užívaly 24 denní režim.
Daylette je kombinované perorální kontraceptivum obsahující ethinylestradiol a progestogen drospirenon. V terapeutických dávkách má drospirenon také antiandrogenní a mírné antimineralokortikoidní vlastnosti. Nemá estrogenní, glukokortikoidní ani antiglukokortikoidní aktivitu. Toto dává drospirenonu farmakologický profil výrazně se podobající přirozenému hormonu progesteronu.
Z klinických studií vyplynulo, že slabé antimineralokortikoidní vlastnosti kombinace drospirenon/ethinylestradiol mají za následek mírný antimineralokortikoidní účinek.
Účinnost a bezpečnost kombinace drospirenon/ethinylestradiol v léčbě středně závažného akné u žen byla hodnocena ve dvou multicentrických, dvojitě slepých, randomizovaných a placebem kontrolovaných studiích v délce 6 měsíců. Ve srovnání s placebem přinesla kombinace drospirenonu a ethinylestradiolu statisticky významné snížení výskytu zánětlivých lézí o 15,6% (49,3 % vs. 33,7 %), nezánětlivých lézí o 18,5% (40,6 % vs. 22,1 %), a lézí celkem o 16,5% (44,6 % vs. 28,1 %). Navíc bylo i o 11,8% (18,6 % vs 6,8 %) vyšší procento pacientek s hodnocením „čistá“ nebo „téměř čistá“ na stupnici „Investigators Static Global Assessment„ (ISGA).
5.2 Farmakokinetické vlastnosti
Drospirenon
Absorpce
Perorálně podaný drospirenon je rychle a téměř úplně absorbován. Maximální koncentrace léčivé látky v séru – asi 38 ng/ml – je dosaženo přibližně za 1 – 2 hodiny po jednorázovém podání. Biologická dostupnost je 76 – 85 %. Na biologickou dostupnost drospirenonu nemá současné požití potravy žádný vliv.
Distribuce
Po perorálním podání klesá sérová hladina drospirenonu s konečným poločasem 31 hodin. Drospirenon je vázán na sérový albumin, není vázán na pohlavní hormony vážící globulin (SHBG) ani na kortikosteroidy vážící globulin (CBG). Jen 3 – 5 % celkové sérové koncentrace léčivé látky je přítomno ve formě volného steroidu. Ethinylestradiolem indukované zvýšení SHBG neovlivňuje vazbu drospirenonu na sérové proteiny. Průměrný distribuční objem drospirenonu je 3,7 ± 1,2 l/kg.
Biotransformace
Drospirenon je po perorálním podání kompletně metabolizován. Nejdůležitějšími metabolity v plasmě jsou kyselá forma drospirenonu vzniklá otevřením laktonového kruhu a 4,5-dihydro-drospirenon-3sulfát, oba vznikají bez účasti systému P450. Drospirenon je in vitro z malé části metabolizován cytochromem P450 3A4 a prokázal schopnost inhibovat tento enzym a dále cytochrom P450 1A1, cytochrom P450 2C9 a cytochrom P450 2C19.
Eliminace
Rychlost metabolické clearance drospirenonu ze séra je 1,5 ± 0,2 ml/min/kg. Drospirenon je vylučován pouze ve stopovém množství v nezměněné formě. Jeho metabolity jsou vylučovány stolicí a močí v poměru 1,2 až 1,4. Poločas vylučování metabolitů močí a stolicí je asi 40 hodin.
Rovnovážný stav
Během léčebného cyklu je maximální koncentrace drospirenonu v rovnovážném stavu okolo 70 ng/ml dosaženo přibližně po 8 dnech léčby. Hladiny drospirenonu se akumulují přibližně faktorem 3 v důsledku poměru konečného poločasu a dávkovacího intervalu.
Zvláštní skupiny pacientů
Vliv poruchy funkce ledvin
Sérové hladiny drospirenonu v rovnovážném stavu u žen s mírnou poruchou funkce ledvin (clearance kreatininu CLcr, 50–80 ml/min) byly srovnatelné s ženami, jejichž funkce ledvin byla normální. U žen se středně závažnou poruchou funkce ledvin (Clcr 30–50 ml/min) byly sérové hladiny drospirenonu v průměru o 37% vyšší ve srovnání s ženami s normální funkcí ledvin. Léčba drospirenonem u žen s mírnou až středně těžkou poruchou funkce ledvin byla také dobře tolerována. Léčba drospirenonem nevykazovala žádný klinicky významný účinek na koncentrace draslíku v séru.
Vliv poruchy funkce jater
U dobrovolnic se středně závažnou poruchou funkce jater byl ve studii hodnotící podání jednotlivé dávky pozorován 50%ní pokles perorální clearance (CL/F) ve srovnání s ženami s normální funkcí jater. Pozorovaný pokles clearance drospirenonu u dobrovolnic se středně závažnou poruchou funkce jater ve srovnání se zdravými se neprojevil jako patrný rozdíl koncentrací draslíku v séru u těchto dvou skupin dobrovolnic. Dokonce ani za přítomnosti diabetes a konkomitantní léčby spironolaktonem (dvou faktorů, které predisponují pacienta k hyperkalemii), nebyl pozorován vzestup draslíku nad hranici rozmezí normálních hodnot. Závěrem lze říci, že u pacientek s mírnou nebo středně závažnou poruchou funkce jater (Child-Pugh B) je drospirenon dobře tolerován.
Etnické skupiny
Nebyly pozorovány žádné klinicky významné rozdíly farmakokinetiky drospirenonu nebo ethinylestradiolu mezi japonskými a kavkazskými ženami.
Ethinylestradiol
Absorpce
Perorálně podaný ethinylestradiol je rychle a kompletně absorbován. Nejvyšší sérové koncentrace okolo 33 pg/ml je dosaženo po jednorázové perorální dávce během 1 – 2 hodin. Absolutní biologická dostupnost je přibližně 60 % následkem presystémové konjugace a first-pass metabolismu. Současné požití stravy snížilo biologickou dostupnost ethinylestradiolu asi u 25% sledovaných subjektů, zatímco u ostatních nebyly pozorovány žádné změny.
Distribuce
Sérové koncentrace ethinylestradiolu klesají ve dvou fázích, konečná dispoziční fáze je charakterizována poločasem přibližně 24 hodin. Ethinylestradiol je vysoce, ale nespecificky vázán na sérový albumin (přibližně 98,5 %) a indukuje vzestup sérové koncentrace SHBG a kortikoid vázajícího globulinu (CBG). Distribuční objem je uváděn kolem 5 l/kg.
Biotransformace
Ethinylestradiol podléhá presystémové konjugaci jak ve stěně tenkého střeva, tak v játrech. Ethinylestradiol je primárně metabolizován aromatickou hydroxylací, vzniká však velké množství různých hydroxylovaných a methylovaných metabolitů, které jsou přítomny jako volné metabolity nebo jako konjugáty s glukuronidy a sulfáty. Metabolická clearance ethinylestradiolu je uváděna kolem 5 ml/min/kg.
Eliminace
Ethinylestradiol není ve významném množství vylučován v nezměněné formě. Metabolity ethinylestradiolu jsou vylučovány močí a žlučí v poměru 4 : 6. Poločas exkrece metabolitů je asi 1 den.
Rovnovážný stav
Rovnovážného stavu je dosaženo během druhé poloviny léčebného cyklu a sérové hladiny ethinylestradiolu se kumulují faktorem od 2,0 do 2,3.
5.3 Předklinické údaje vztahující se k bezpečnosti
U laboratorních zvířat byly účinky drospirenonu a ethinylestradiolu omezeny na ty, které jsou spojeny s jejich známým farmakologickým působením. Zejména studie reprodukční toxicity odhalily u zvířat embryotoxické a fetotoxické účinky, které jsou považovány za druhově specifické. Při expozici v dávkách vyšších, než jaké jsou přijímány u uživatelek kombinace drospirenon/ethinylestradiol, byly pozorovány účinky na diferenciaci pohlaví u plodů potkanů, ale nikoli u plodů opic.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety (aktivní):
Monohydrát laktosy
Kukuřičný škrob
Předbobtnalý kukuřičný škrob
Potahová soustava kollicoat IR Magnesium-stearát
Obal tablety (aktivní):
Polyvinylalkohol
Oxid titaničitý (E171)
Mastek
Makrogol 3350
Sójový lecithin
Jádro tablety (placebo):
Mikrokrystalická celulosa
Laktosa
Předbobtnalý kukuřičný škrob Magnesium-stearát
Koloidní bezvodý oxid křemičitý
Obal tablety (placebo):
Polyvinylalkohol
Oxid titaničitý (E171)
Makrogol 3350
Mastek
Hlinitý lak indigokarmínu (E132)
Hlinitý lak chinolinové žluti (E104)
Černý oxid železitý (E172)
Hlinitý lak oranžové žluti (E110)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
-
2 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25°C. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Přípravek Daylette 3 mg/0,02 mg potahované tablety je balen v bezbarvých průhledných PVC/PE/PVDC-Al blistrech. Blistry jsou zabaleny v papírové krabičce s příbalovou informací a v každé krabičce je pouzdro, na kterém jsou nalepeny etikety s textem v portugalštině. Součástí balení jsou 3 volně vložené etikety s českými zkratkami názvů dnů v týdnu.
Velikost balení:
-
3 x (24+4) potahovaných tablet
Text na blistru a na kartonovém pouzdře je v portugalštině. Překlad textu je uveden v Příbalové informaci.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Žádné zvláštní požadavky.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Gedeon Richter Plc.
Gyómrói út 19–21.
1103 Budapešť,
Maďarsko
Kabu Pharma s.r.o., U Staré tvrze 285/21, Třeboradice, 196 00 Praha 9, Česká republika
8.
REGISTRAČNÍ ČÍSLO
17/070/11-C /PI/003/14
Další informace o léčivu DAYLETTE 3 MG/0,02 MG POTAHOVANÉ TABLETY
Jak
se DAYLETTE 3 MG/0,02 MG POTAHOVANÉ TABLETY
podává: perorální podání - potahovaná tableta
Výdej
léku: na lékařský předpis
Balení: Blistr
Velikost
balení: 3X28
Držitel rozhodnutí o registraci daného léku v České republice:
Gedeon Richter Plc., Budapešť
E-mail: info@richtergedeon.cz
Telefon: 261141200