Souhrnné informace o léku - ROSEMIG SPRINTAB 100 MG
sp.zn.sukls156419/2014
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Rosemig Sprintab 50 mg
Rosemig Sprintab 100 mg dispergovatelné tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Rosemig Sprintab 50 mg: jedna dispergovatelná tableta obsahuje sumatriptani succinas 70,0 mg, což odpovídá sumatriptanum 50,0 mg.
Rosemig Sprintab 100 mg: jedna dispergovatelná tableta obsahuje sumatriptani succinas 140,0 mg, což odpovídá sumatriptanum 100,0 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Dispergovatelné tablety
Rosemig Sprintab 50 mg: růžové trojúhelníkové bikonvexní filmem potažené dispergovatelné tablety, z jedné strany vyraženo „GS 1YM“, z druhé strany „50“.
Rosemig Sprintab 100 mg: bílé trojúhelníkové bikonvexní filmem potažené dispergovatelné tablety, z jedné strany vyraženo „GS YE7“, z druhé strany „100“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Rosemig Sprintab je indikován k okamžité úlevě při migrenózním záchvatu jak s aurou, tak bez ní, včetně akutní léčby migrenózního záchvatu souvisejícího s menstruací u žen.
4.2 Dávkování a způsob podání
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Rosemig Sprintab 50 mg
Rosemig Sprintab 100 mg dispergovatelné tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Rosemig Sprintab 50 mg: jedna dispergovatelná tableta obsahuje sumatriptani succinas 70,0 mg, což odpovídá sumatriptanum 50,0 mg.
Rosemig Sprintab 100 mg: jedna dispergovatelná tableta obsahuje sumatriptani succinas 140,0 mg, což odpovídá sumatriptanum 100,0 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Dispergovatelné tablety
Rosemig Sprintab 50 mg: růžové trojúhelníkové bikonvexní filmem potažené dispergovatelné tablety, z jedné strany vyraženo „GS 1YM“, z druhé strany „50“.
Rosemig Sprintab 100 mg: bílé trojúhelníkové bikonvexní filmem potažené dispergovatelné tablety, z jedné strany vyraženo „GS YE7“, z druhé strany „100“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Rosemig Sprintab je indikován k okamžité úlevě při migrenózním záchvatu jak s aurou, tak bez ní, včetně akutní léčby migrenózního záchvatu souvisejícího s menstruací u žen.
4.2 Dávkování a způsob podání
Sumatriptan se neužívá profylakticky. Doporučená dávka sumatriptanu nemá být překračována.
Sumatriptan se doporučuje k akutní léčbě migrenózního záchvatu v monoterapii. Nesmí se podávat společně s ergotaminem ani deriváty ergotaminu (včetně methysergidu) (viz bod 4.3).
Doporučuje se podat sumatriptan co nejdříve po nástupu migrenózních bolestí hlavy, ale je stejně účinný, je-li podán v kterémkoliv stádiu záchvatu.
Doporučená perorální dávka sumatriptanu ve formě dispergovatelných tablet je 50mg tableta. Někteří pacienti však mohou potřebovat dávku 25 mg (musí se použít jiný přípravek) nebo100 mg.
Nedojde-li po první dávce sumatriptanu ke zlepšení obtíží, nemá být podána druhá dávka v průběhu téhož záchvatu. V těchto případech lze podat paracetamol, kyselinu acetylsalicylovou nebo nesteroidní antirevmatika. Sumatriptan lze podat v průběhu následujících záchvatů.
Pokud se po přechodném zlepšení po první dávce znovu objeví obtíže (rekurence ataky), lze podat další dávku při dodržení minimálního intervalu dvou hodin mezi dávkami a za předpokladu, že se nepodá více než 300 mg léčivé látky v průběhu 24 hodin.
Tablety se polykají celé a zapíjejí se vodou. Pacienti, kteří mají potíže s polykáním, mohou před podáním tabletu rozpustit v malém množství vody. Rosemig Sprintab má po rozpuštění hořkou chuť.
Účinnost a bezpečnost dispergovatelných tablet sumatriptanu nebyla u dětí mladších 10 let stanovena. Pro tuto věkovou skupinu nejsou k dispozici žádné klinické údaje.
Účinnost a bezpečnost dispergovatelných tablet sumatripatnu u dětí nebyla v klinických studiích provedených u této věkové skupiny prokázána (viz bod 5.1).
Zkušenosti s použitím standardních tablet sumatriptanu u nemocných nad 65 let jsou omezené. Farmakokinetika se však významně neliší od mladší populace. Dokud nebudou k dispozici další klinické údaje, podávání sumatriptanu ve formě dispergovatelných tablet se nemocným starším než 65 let nedoporučuje.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku.
Sumatriptan se nesmí podávat pacientům, kteří prodělali infarkt myokardu, nebo trpí ischemickou chorobou srdeční (ICHS), koronárními vazospazmy (Prinzmetalovou variantní formou anginy pectoris), onemocněním periferních cév ani pacientům se subjektivními nebo objektivními příznaky odpovídajícími ischemické chorobě srdeční.
Sumatriptan se nesmí podávat pacientům s anamnézou cévní mozkové příhody (CMP) ani tranzitorní ischemické ataky (TIA).
Kontraindikované je podání sumatriptanu pacientům se středně závažnou a závažnou hypertenzí a pacientům s mírnou nekontrolovanou hypertenzí.
Sumatriptan se nesmí podávat pacientům s těžkou poruchou funkce jater.
Současné podávání s ergotaminem nebo deriváty ergotaminu (včetně methysergidu) nebo s jakýmkoli triptanem/agonistou 5-hydroxytryptamin-1 receptoru (5HT1) je kontraindikované (viz bod 4.5).
Současné podávání sumatriptanu a inhibitorů monoaminooxidázy (MAO) je kontraindikováno. Sumatriptan se nesmí podávat v průběhu dvou týdnů po ukončení léčby přípravkem obsahujícím inhibitory MAO.
4.4 Zvláštní upozornění a opatření pro použití
Sumatriptan se má podávat pouze při jasné diagnóze migrény.
Sumatriptan není indikován k léčbě pacientů s hemiplegickou, bazilární ani oftalmoplegickou formou migrény.
Před zahájením léčby sumatriptanem má být pacientům s netypickými příznaky nebo pacientům, u kterých nebyla dříve stanovena diagnóza migrény, věnována pozornost, aby byla vyloučena jiná potenciálně závažná neurologická onemocnění (jako např. cévní mozková příhoda, tranzitorní ischemická ataka).
Podávání sumatriptanu může být spojováno s výskytem tranzitorních symptomů včetně bolesti na hrudi a pocitu svírání, které mohou být intenzivní a mohou se šířit až do krku (viz bod 4.8). Tam, kde tyto příznaky vedou k podezření na ischemickou chorobu srdeční, nemají být podávány další dávky sumatriptanu a pacient má absolvovat odpovídající kardiologické vyšetření.
Sumatriptan se nemá podávat pacientům s rizikovými faktory ischemické choroby srdeční, včetně těch pacientů, kteří jsou silnými kuřáky, nebo podstupují substituční léčbu nikotinem, bez předchozího kardiovaskulárního vyšetření (viz bod 4.3). Zvláštní opatrnost je zapotřebí při podávání ženám po menopauze a mužům nad 40 let s těmito rizikovými faktory. Avšak ani tato vyšetření nemusí zachytit všechny pacienty s kardiálním onemocněním, takže ve velmi vzácných případech se mohou závažné kardiální příhody vyskytnout u pacientů bez základního kardiovaskulárního onemocnění (viz bod 4.8).
Sumatriptan má být podáván s opatrností pacientům s mírnou kontrolovanou hypertenzí, protože u malého počtu pacientů bylo pozorováno přechodné zvýšení krevního tlaku a periferní vaskulární rezistence (viz bod 4.3).
Po uvedení přípravku na trh byl u pacientů užívajících současně selektivní inhibitory zpětného vychytávání serotoninu (SSRI) a sumatriptan v ojedinělých případech popsán výskyt serotoninového syndromu (včetně mentální alterace, autonomní nestability a neuromuskulárních abnormalit). Serotoninový syndrom byl hlášen při současné léčbě triptany a inhibitory zpětného vychytávání serotoninu a noradrenalinu (SNRI).
Je-li současné podávání sumatriptanu a SSRI/SNRI klinicky nezbytné, je třeba pacienty odpovídajícím způsobem sledovat (viz bod 4.5).
Sumatriptan by měl být podáván s opatrností nemocným s onemocněními, která mohou výrazně ovlivnit vstřebávání, metabolismus, nebo vylučování léků, např. s poruchou jaterních nebo renálních funkcí (Child-Pugh stupně A nebo B, viz bod 5.2).
Sumatriptan by měl být podáván s opatrností pacientům s anamnézou epileptických záchvatů nebo jiných rizikových faktorů, které snižují práh pro vznik záchvatů, protože v souvislosti se sumatriptanem byly hlášeny epileptické záchvaty (viz bod 4.8).
U pacientů se známou přecitlivělostí na sulfonamidy může po podání sumatriptanu dojít k alergické reakci, a to od projevů kožní přecitlivělosti až po anafylaktickou reakci.
Údaje o výskytu zkřížené senzitivity se sulfonamidy jsou omezené. Při zahájení léčby sumatriptanem je však nutné mít tuto skutečnost na paměti.
Při současném podání triptanů a rostlinných přípravků obsahujících třezalku tečkovanou (Hypericum perforatum) se nežádoucí účinky mohou vyskytnout častěji.
Dlouhodobé užívání jakéhokoli léku k léčbě bolestí hlavy může tyto bolesti zhoršovat. Pokud k této situaci dojde, nebo je na ní podezření, je třeba vyhledat lékařskou pomoc a léčbu ukončit. Diagnóza bolesti hlavy z nadužívání léků (MOH – medication overuse headache) se nabízí v případě, kdy pacienti mají časté nebo denní bolesti hlavy i když (nebo protože) pravidelně užívají medikaci k léčbě bolesti hlavy.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nejsou důkazy o vzájemných interakcích s propranololem, flunarizinem, pizotifenem nebo alkoholem.
Údaje týkající se interakcí s přípravky obsahujícími ergotamin nebo jiné triptany/agonisty 5-hydroxytryptamin-1 receptoru (5-HT1) jsou omezené. Zvýšené riziko vzniku koronárních vazospazmů je teoreticky možné, proto je souběžné podávání těchto přípravků kontraindikováno (viz bod 4.3).
Doba, která by měla uplynout mezi užitím sumatriptanu a přípravků obsahujících ergotamin, nebo jiné triptany/agonisty 5-hydroxytryptamin-1 receptoru (5-HT1), není známa. Toto bude také záviset na dávkách a typu použitého přípravku. Účinky se mohou sčítat. Doporučuje se počkat alespoň 24 hodin po podání přípravků obsahujících ergotamin nebo jiné triptany/agonisty 5-hydroxytryptamin-1 receptoru (5-HT1) před podáním sumatriptanu. Naopak po podání sumatriptanu se doporučuje počkat alespoň šest hodin před podáním přípravků obsahujících ergotamin a alespoň 24 hodin před podáním jiných triptanů/agonistů 5-hydroxytryptamin-1 receptoru (5-HT1).
Mezi sumatriptanem a inhibitory MAO může dojít k vzájemným interakcím, proto je jejich současné podávání kontraindikované (viz bod 4.3).
V postmarketingových hlášeních u pacientů užívajících současně selektivní inhibitory zpětného vychytávání serotoninu (SSRI) a sumatriptan byl v ojedinělých případech popsán výskyt serotoninového syndromu (včetně mentální alterace, autonomní nestability a neuromuskulárních abnormalit). Serotoninový syndrom byl rovněž hlášen při současné léčbě triptany a SNRI (viz bod 4.4).
Vzhledem k možnosti vzniku interakcí se doporučuje přerušit terapii přípravky obsahujícími třezalku tečkovanou.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Postmarketingové údaje získané z mnoha prospektivních registrů těhotenství dokumentují výsledky od více než 1000 těhotných žen užívajících sumatriptan během prvního trimestru. Ačkoliv jsou k dispozici pouze nedostatečné údaje k vyvození definitivních závěrů, nálezy neprokázaly zvýšený výskyt vrozených defektů, ani konzistentní vzorec vrozených defektů u žen vystavených působení sumatriptanu ve srovnání s celkovou populací. Zkušenosti s podáváním sumatriptanu během druhého a třetího trimestru jsou omezené.
Předklinické studie provedené na zvířatech neprokázaly přímý teratogenní účinek nebo škodlivý vliv na peri- a postnatální vývoj. Přesto, embryofetální životaschopnost může být ovlivněna u králíků (viz bod 5.3).
Sumatriptan by měl být v těhotenství podán pouze tehdy, převáží-li očekávaný přínos pro matku možná rizika pro plod.
Kojení
Bylo zjištěno, že po subkutánním podání sumatriptan přechází do mateřského mléka. Pro minimalizaci expozice kojence se doporučuje kojit až za 12 hodin po jeho podání. Během této doby je nutné mléko odstříkat a znehodnotit.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit a obsluhovat stroje nebyly provedeny.
Může se vyskytnout ospalost buď jako důsledek migrény anebo po podání sumatriptanu, která může ovlivnit schopnost řídit a obsluhovat stroje.
Pacientům vykonávajícím činnosti vyžadující zvýšenou pozornost, soustředění a koordinaci pohybů (jako je např. řízení nebo obsluha strojů) se doporučuje opatrnost.
4.8 Nežádoucí účinky
Nežádoucí účinky jsou uvedeny níže podle tříd orgánových systémů a frekvence výskytu. Četnost je definována: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
Některé příznaky hlášené jako nežádoucí účinky mohou souviset s příznaky migrény.
Poruchy imunitního systému
| Není známo: | Hypersenzitivní reakce v rozsahu od kožních reakcí přecitlivělosti (jako např. kopřivka) až po anafylaktickou reakci. |
| Poruchy nervového systému | |
| Časté: | Závrať, ospalost, smyslové poruchy včetně parestézie a hypestézie. |
| Není známo: | Epileptické záchvaty. Ačkoliv se některé vyskytly u pacientů buď s anamnézou epileptických záchvatů, nebo v souvislosti se stavy predisponujícími ke vzniku epileptických záchvatů, byly rovněž hlášeny u pacientů bez zjevných predisponujících faktorů. Třes, dystonie, nystagmus, skotomy. |
| Poruchy oka | |
| Není známo: | Záblesky před očima, dvojité vidění, snížení vizu. Ztráta vizu (obvykle přechodná) včetně hlášení o trvalých defektech. Avšak poruchy vizu mohou rovněž doprovázet vlastní záchvat migrény. |
| Srdeční poruchy | |
| Není známo: | Bradykardie, tachykardie, palpitace, arytmie, přechodné ischemické změny na EKG, koronární arteriální vazospasmus, angina pectoris, infarkt myokardu (viz bod 4.3 a 4.4). |
| Cévní poruchy | |
| Časté: | Přechodné zvýšení krevního tlaku objevující se brzy po podání léčby. Zčervenání. |
| Není známo: | Hypotenze, Raynaudův fenomén. |
Respirační, hrudní a mediastinální poruchy
| Časté: | Dyspnoe. |
Gastrointestinální poruchy
| Časté: | Nauzea a zvracení u některých pacientů, není však zřejmé, zda souvisí s podáním sumatriptanu nebo základním onemocněním. |
| Není známo: | Ischemická kolitida, průjem. |
Poruchy svalové a kosterní soustavy a pojivové tkáně
| Časté: | Pocit těžkosti (tento příznak je obvykle přechodný, může být intenzivní a může postihnout kteroukoliv část těla včetně hrudníku a krku). Bolest svalů. |
| Není známo: | Ztuhlost šíje, artralgie. |
Celkové poruchy a reakce v místě aplikace
| Časté: | Bolest, pocit tepla nebo chladu, tlaku nebo napětí (tyto příznaky jsou obvykle přechodné, mohou být intenzívní a mohou postihnout kteroukoliv část těla včetně hlavy a krku). Pocit slabosti, únava (oba příznaky jsou většinou mírné až středně závažné a přechodné). |
Vyšetření
| Velmi vzácné: | Občas byly pozorovány mírné poruchy jaterních funkčních testů. |
Není známo:
Úzkost.
Není známo:
Hyperhidróza.
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
Známky a příznaky
Podávání perorálních dávek do 100 mg nebylo spojeno s výskytem jiných než výše uvedených nežádoucích účinků.
Léčba
V případě předávkování sumatriptanem má být pacient monitorován po dobu alespoň 10 hodin a podle potřeby má být zahájena standardní podpůrná léčba.
Není známo, jaký účinek na koncentraci sumatriptanu v plazmě má hemodialýza nebo peritoneální dialýza.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antimigrenikum, selektivní agonista 5-HT1 receptorů. ATC kód: N02CC01
Mechanismus účinku
Sumatriptan je specifický selektivní agonista 5-hydroxytryptamin-1 receptoru (5HT1D) bez účinku na další subtypy 5HT-receptorů (5HT2–7). Receptor 5HT1D se nachází převážně v kraniálních cévách a zprostředkovává vazokonstrikci.
U zvířat sumatriptan způsobuje selektivní vazokonstrikci v karotickém řečišti, které zásobuje krví extrakraniální a intrakraniální tkáně, např. mozkomíšní pleny, ale neovlivňuje průtok krve mozkem. Rozšíření a/nebo edém těchto cév se zdají být základním mechanismem vzniku migrény u člověka.
Dosavadní výzkumy navíc nasvědčují, že sumatriptan inhibuje aktivitu n. trigeminus. Oba tyto vlivy se mohou podílet na antimigrenózním účinku sumatriptanu u člověka.
Farmakodynamické účinky
Klinická odpověď přichází 10 až 15 min po podkožně aplikované 6mg injekci, 15 minut po dávce 20 mg podané intranazálně a přibližně 30 minut po perorálním podání standardní 100mg tablety nebo po podání 25 mg rektálně.
Při podání 50 mg a 100 mg dispergovatelných tablet došlo ke zmírnění bolesti u malého počtu jedinců již za 30 minut resp. 20 minut a procento pacientů reagujících na léčbu se zvyšovalo v průběhu dvou hodin až na 67 % resp. 72 % jedinců, ve srovnání se 42 % jedinců užívajících placebo. K odeznění bolesti došlo u malého počtu jedinců již za 33 minut resp. 26 minut a procento pacientů bez bolesti se v průběhu dvou hodin zvyšovalo až na 40 % resp. 47 %, ve srovnání s 15 % jedinců užívajících placebo (viz bod 5.2).
I když je doporučená dávka pro perorální podání sumatriptanu 50 mg, liší se závažnost migrenózních záchvatů u jednotlivých pacientů i mezi nimi. V klinických studiích se prokázalo, že dávky mezi 25 až 100 mg mají větší účinek než placebo, ale dávka 25 mg má statisticky signifikantně menší účinek než dávky 50 a 100 mg.
Sumatriptan je účinný při akutní léčbě migrény, včetně migrény související s menstruací.
Sumatriptan, ve formě dispergovatelných tablet nebyl hodnocen u dospívajících. Několik placebem kontrolovaných klinických studií hodnotilo bezpečnost a účinnost perorálně podávaného sumatriptanu u zhruba 800 dětí a dospívajících s migrénou ve věku 10 až 17 let. Tyto studie neprokázaly relevantní rozdíly ve zmírnění bolestí hlavy po dvou hodinách mezi placebem a jakoukoli dávkou sumatriptanu. Profil nežádoucích účinků perorálně podaného sumatriptanu u dospívajících ve věku 10 až 17 let byl podobný, jako profil zaznamenaný ze studií u dospělých pacientů.
5.2 Farmakokinetické vlastnosti
Nezdá se, že by migrenózní záchvaty významně ovlivňovaly farmakokinetiku perorálně podaného sumatriptanu.
Absorpce
Po 100mg dávce je průměrná maximální koncentrace v plazmě 54 ng/ml.
Průměrná absolutní perorální biologická dostupnost je 14 %, zčásti kvůli presystémovému metabolismu a zčásti kvůli neúplnému vstřebání.
Po perorálním podání dispergovatelných tablet s tučným jídlem se Cmax sumatriptanu zvyšuje o 15 %.
Distribuce
Vazba na plazmatické proteiny je nízká (14 až 21 %), průměrný distribuční objem je 170 litrů.
Metabolismus
Hlavní metabolit, indolacetátový analog sumatriptanu, se vylučuje zejména močí, kde je přítomen jako volná kyselina a glukuronidový konjugát. Nemá žádné známé účinky na aktivitu receptorů 5HT1 či 5HT2. Sekundární metabolity nebyly identifikovány.
Eliminace
Poločas vylučování je přibližně 2 hodiny. Celková průměrná plazmatická clearance je přibližně 1160 ml/min a průměrná renální plazmatická clearance je asi 260 ml/min.
Extrarenální clearance se podílí asi 80 % na celkové clearance a naznačuje, že sumatriptan se primárně metabolizuje oxidačními mechanismy pomocí monoaminooxidázy A.
Zvláštní skupiny pacientů
Porucha funkce jater
Po perorálním podání dojde u pacientů s poruchou funkce jater ke snížení presystémové clearance sumatriptanu, které vede ke zvýšení jeho plazmatické koncentrace (viz bod 4.4).
Nástup účinku sumatriptanu 50 mg a 100 mg, dispergovatelných tablet u dospělých byl prezentován ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích stejného designu. Údaje z těchto studií byly sloučeny k získání jednoho výsledku pro každý cílový parametr. Celkově 2696 jedinců se středně závažnou až závažnou formou migrénozní bolesti uvádělo čas do ústupu bolesti a období bez bolesti ve skupinách užívajících sumatriptan 50 mg, 100 mg a placebo. Křivky nástupu úlevy od bolesti (definován jako snížení závažnosti bolesti od středně závažné nebo závažné bolesti do mírné nebo bez bolesti) byly vytvořeny pro sumatriptan a placebo pro období dvou hodin po podání léčby. Začátek ústupu bolesti byl definován jako nejranější časový okamžik, kdy bylo poprvé dosaženo statistické významnosti, ve srovnání s placebem, která byla udržována v celém následujícím časovém období křivky od 0 do 2 hodin. Období bez bolesti (definováno jako snížení závažnosti bolesti od závažné nebo středně závažné bolesti do stavu bez bolesti) byl hodnocen za použití podobných metod (viz bod 5.1).
Procento jedinců dosahujících úlevy bolesti (obrázek 1), nebo bez bolesti (obrázek 2) během dvou hodin po podání léčby bylo významně vyšší mezi jedinci užívajícími sumatriptan (dispergovatelné tablety 50 mg nebo 100 mg), ve srovnání s těmi jedinci, kteří užívali placebo (p<0,001).
Obrázek 1: Čas do úlevy od bolesti až do dvou hodin po podání léčby*
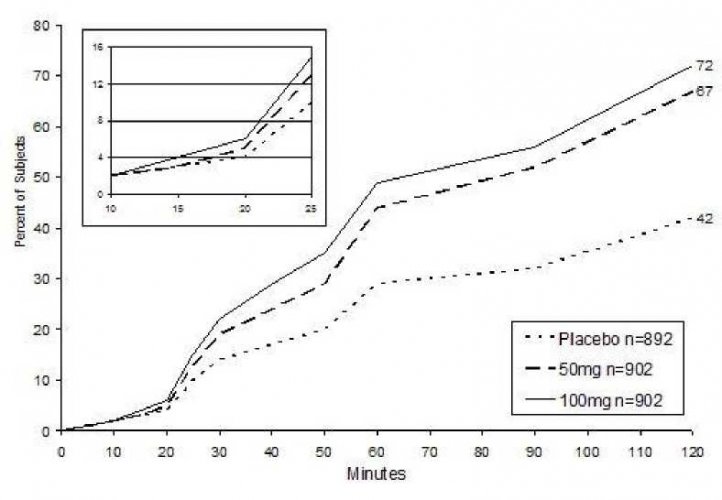
Percent of Subjects: procento subjektů, minutes: minuty
Kaplan-Meierovo znázornění je založené na kombinaci údajů získaných ze dvou studií, kdy každá z nich prokázala účinnost léčby. Vložený obrázek v obrázku 1 ukazuje procento subjektů s úlevou od bolesti během prvních 10 – 25 minut po podání léčby.
Při podání 50 mg a 100 mg dispergovatelných tablet byla doba do nástupu úlevy od bolesti 30 minut, resp. 20 minut, na základě kombinovaných údajů. Od tohoto okamžiku se procento respondérů zvyšovalo až na 67 % , resp. 72 % jedinců, kteří dosáhli úlevy za 2 hodiny po podání sumatriptanu v dávce 50 mg resp. 100 mg, ve srovnání se 42 % jedinců ve skupině s placebem (viz obrázek 1).
Obrázek 2: Čas do období bez bolesti až do 2 hodin po podání léčby
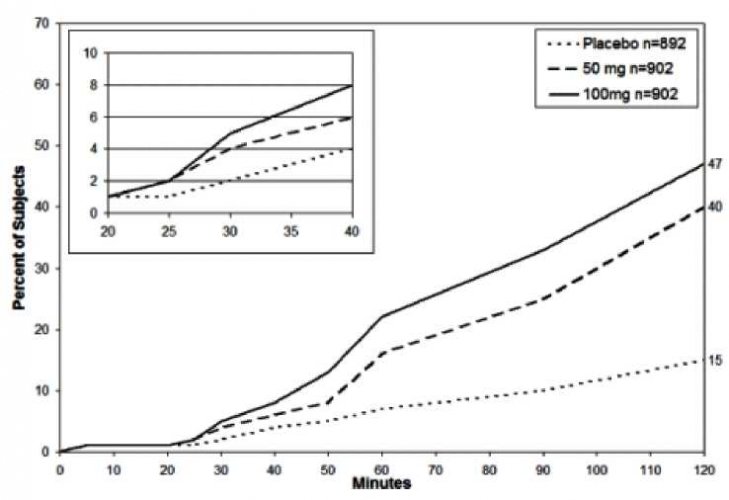
Percent of Subjects: procento subjektů, minutes: minuty
*Kaplan-Meierovo znázornění je založené na kombinaci údajů získaných ze dvou studíích, kdy každá z nich prokázala účinnost léčby. Vložený obrázek v obrázku 1 ukazuje procento subjektů s ústupem bolesti během prvních 20 – 40 minut po podání léčby.
Doba do nástupu období bez bolesti po podání dispergovatelných tablet sumatriptanu o síle 50 mg byla 33 minut a po 100 mg 26 minut na základě kombinovaných údajů. Od tohoto okamžiku se procento jedinců bez bolesti zvyšovalo až na 40 % resp. 47 % za dvě hodiny ve srovnání s 15 % jedinců ve skupině s placebem (viz obrázek 2).
5.3 Předklinické údaje vztahující se k bezpečnosti přípravku
Kancerogenita, mutagenita
V průběhu studií in vitro a studií na zvířatech nebyla prokázána genotoxicita a kancerogenita.
Reprodukční toxicita
Ve studiích fertility u potkanů byl pokles úspěšné inseminace pozorován při perorálně podaném sumatriptanu u plazmatických hladin přibližně 200krát vyšších než hladin u člověka po perorální dávce 100 mg.
Tento účinek se neobjevil ve studiích se subkutánně podaným přípravkem, kde maximální plazmatické hladiny dosáhly přibližně 150násobku hladin pozorovaných u člověka po perorálním podání.
Těhotenství a kojení
U potkanů ani králíků nebyl prokázán teratogenní účinek a sumatriptan nepůsobil na postnatální vývoj potkanů.
Když byl sumatriptan podáván březím samicím králíka během období organogeneze, ojediněle vedl k úmrtí plodu při dávkách dostatečně vysokých, aby vedly k maternální toxicitě.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Rosemig Sprintab 50 mg:
hydrogenfosforečnan vápenatý, mikrokrystalická celulosa, hydrogenuhličitan sodný, sodná sůl kroskarmelosy, magnesium-stearát
potahová soustava Opadry YS-1–1441G růžová: hypromelosa, oxid titaničitý (E171), triacetin, červený oxid železitý (E172).
Rosemig Sprintab 100 mg:
hydrogenfosforečnan vápenatý, mikrokrystalická celulosa, hydrogenuhličitan sodný, sodná sůl kroskarmelosy, magnesium-stearát
potahová soustava Opadry OY-S-7322 bílá: hypromelosa, oxid titaničitý (E171), triacetin.
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
-
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
6.5 Druh obalu a velikost balení
Blistr (PA-Al-PVC/Al nebo PA-Al-PVC/Al/papír), krabička.
Velikosti balení:
2 × 50 mg,
-
4 × 50 mg,
6 × 50 mg,
2 × 100 mg,
4 × 100 mg,
6 × 100 mg.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Montrose Fine Chemical Company Limited,
Irvine, Ayrshire,
Velká Británie
8. REGISTRAČNÍ ČÍSLA
Rosemig Sprintab 50 mg
33/195/04-C
Rosemig Sprintab 100 mg
33/196/04-C
Další informace o léčivu ROSEMIG SPRINTAB 100 MG
Jak
se ROSEMIG SPRINTAB 100 MG
podává: perorální podání - dispergovatelná tableta
Výdej
léku: na lékařský předpis
Balení: Blistr
Velikost
balení: 4 I
Držitel rozhodnutí o registraci daného léku v České republice:
Montrose Fine Chemical Company Ltd., Irvine
E-mail: cz.info@gsk.com
Telefon: 222 001 111