Souhrnné informace o léku - EZETIMIB/SIMVASTATIN MYLAN
1. NÁZEV PŘÍPRAVKU
Ezetimib/Simvastatin Mylan 10 mg/10 mg tablety
Ezetimib/Simvastatin Mylan 10 mg/20 mg tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta 10 mg/10 mg obsahuje ezetimibum 10 mg a simvastatinum 10 mg.
Jedna tableta 10 mg/20 mg obsahuje ezetimibum 10 mg a simvastatinum 20 mg.
Pomocné látky se známým účinkem:
Jedna tableta 10 mg/10 mg obsahuje 68,85 mg monohydrátu laktózy.
Jedna tableta 10 mg/20 mg obsahuje 147,71 mg monohydrátu laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tableta.
Ezetimib/Simvastatin Mylan 10 mg/10 mg: bílé až téměř bílé, oválné, bikonvexní tablety velikosti přibližně 9 mm na délku a 4,5 mm na šířku s vyraženým M na jedné straně a ES1 na druhé straně.
Ezetimib/Simvastatin Mylan 10 mg/20 mg: bílé až téměř bílé, oválné, bikonvexní tablety velikosti přibližně 11 mm na délku a 6,5 mm na šířku s vyraženým M na jedné straně a ES2 na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence kardiovaskulárních příhod
Přípravek Ezetimib/Simvastatin Mylan je indikován ke snížení rizika kardiovaskulárních příhod (viz bod 5.1) u pacientů s ischemickou chorobou srdeční (ICHS) a s akutním koronárním syndromem (AKS) v anamnéze, bez ohledu na to, zda předtím byli léčeni statinem.
Hypercholesterolemie
Přípravek Ezetimib/Simvastatin Mylan je indikován jako přídatná terapie k dietě u pacientů s primární (heterozygotní familiární a nefamiliární) hypercholesterolemií nebo smíšenou hyperlipidemií, kde je vhodné použití kombinovaného přípravku:
- u pacientů, kteří nejsou samotným statinem dostatečně kontrolováni
- u pacientů, kteří se již léčí statinem a ezetimibem.
Homozygotní familiární hypercholesterolemie (HoFH)
Přípravek Ezetimib/Simvastatin Mylan je indikován jako přídatná terapie k dietě u pacientů s HoFH. Pacienti mohou dostávat i další přídatnou terapii (např. aferézu nízkodenzitního lipoproteinu [LDL]).
4.2 Dávkování a způsob podání
Text odkazuje i na síly přípravku 10 mg/40 mg a 10 mg/80 mg. Tyto síly nejsou ovšem v České republice pod názvem Ezetimib/Simvastatin Mylan registrovány. Může se ale stát, že budete tyto síly užívat v podobě přípravku od jiného držitele rozhodnutí o registraci, proto i těmto informacím věnujte pozornost.
Dávkování
Hypercholesterolemie
Pacient má být na odpovídající hypolipidemické dietě a v průběhu léčby přípravkem Ezetimib/Simvastatin Mylan má v dietě pokračovat.
Dávkovací rozmezí kombinace ezetimibu a simvastatinu je od 10/10 mg/den až po 10/80 mg/den večer. Všechny velikosti dávky nemusí být k dispozici ve všech členských státech. Běžná dávka je 10/20 mg/den nebo 10/40 mg/den podána jednorázově večer. 10/80mg dávka se doporučuje pouze u pacientů se závažnou hypercholesterolemií a s vysokým rizikem kardiovaskulárních komplikací, u kterých se při nižších dávkách nedosáhlo léčebných cílů a pokud se očekává, že přínosy převáží nad potenciálními riziky (viz body 4.4 a 5.1). Při zahajování léčby nebo úpravě dávky je třeba vzít v úvahu koncentraci cholesterolu nízkodenzitního lipoproteinu (low-density lipoprotein cholesterol, LDL-C), riziko ischemické choroby srdeční a odpověď na aktuální cholesterol snižující léčbu.
Dávku přípravku Ezetimib/Simvastatin Mylan je nutno individuálně upravit podle známé účinnosti různých sil dávek kombinace ezetimibu a simvastatinu (viz bod 5.1, Tabulka 1) a podle odpovědi na aktuální cholesterol snižující léčbu. Úprava dávky, pokud je zapotřebí, se musí provádět s odstupem minimálně 4 týdnů.
Pacienti s ischemickou chorobou srdeční a akutním koronárním syndromem v anamnéze
Ve studii zaměřené na snížení rizika kardiovaskulárních příhod (IMPROVE-IT) byla počáteční dávka 10/40 mg jednou denně večer. Dávka 10/80 mg se doporučuje pouze za předpokladu, že přínosy převáží nad potenciálními riziky.
Homozygotní familiární hypercholesterolemie
Doporučená dávka kombinace ezetimibu a simvastatinu pro pacienty s homozygotní familiární hypercholesterolemií je 10/40 mg/den večer. Dávka 10/80 mg se doporučuje, pouze pokud se předpokládá, že přínosy převáží nad potenciálními riziky (viz dále; body 4.3 a 4.4).
Přípravek Ezetimib/Simvastatin Mylan lze použít jako přídatný přípravek k dalším formám hypolipidemické léčby (např. LDL aferéza) u těchto pacientů, nebo pokud není taková léčba k dispozici.
U pacientů užívajících lomitapid současně s přípravkem Ezetimib/Simvastatin Mylan nesmí dávka kombinace ezetimibu a simvastatinu překročit množství 10/40 mg denně (viz body 4.3, 4.4 a 4.5).
Současné podávání s jinými léčivými přípravky
Přípravek Ezetimib/Simvastatin Mylan je nutno podávat > 2 hodiny před nebo > 4 hodiny po podání sekvestrantu žlučových kyselin.
U pacientů užívajících amiodaron, amlodipin, verapamil, diltiazem nebo přípravky obsahující elbasvir nebo grazoprevir současně s kombinací ezetimibu a simvastatinu nesmí dávka přípravku Ezetimib/Simvastatin Mylan překročit hodnotu 10/20 mg/den (viz body 4.4 a 4.5).
U pacientů, kteří užívají hypolipidemické dávky niacinu (> 1 g/denně) současně s kombinací ezetimibu a simvastatinu, nesmí dávka ezetimibu/simvastatinu překročit hodnotu 10/20 mg/den (viz body 4.4 a 4.5).
Starší pacienti
U starších pacientů není nutno dávku upravovat (viz bod 5.2).
Pediatrická populace
Léčba musí být zahájena pod dohledem specialisty.
Dospívající > 10 let věku (pubertální status: chlapci Tannerův stupeň II a vyšší a dívky, které již alespoň jeden rok menstruují): klinické zkušenosti u pediatrických a dospívajících pacientů (ve věku 10 až 17 let) jsou omezené. Obvyklá doporučená zahajovací dávka je 10/10 mg jednou denně večer. Doporučené dávkové rozmezí je 10/10 až maximálně 10/40 mg/den (viz body 4.4 a 5.2).
Děti < 10 let: kombinace ezetimibu a simvastatinu se u dětí mladších 10 let kvůli nedostatku údajů o bezpečnosti a účinnosti nedoporučuje (viz bod 5.2). Zkušenosti s dětmi před pubertou jsou omezené.
Pacienti s poruchou funkce jater
U pacientů s lehkou poruchou funkce jater (Child-Pughovo skóre 5 až 6) není nutno dávkování upravovat. Nedoporučuje se podávat kombinaci ezetimibu a simvastatinu pacientům se středně těžkou (Child-Pughovo skóre 7 až 9) nebo těžkou poruchou funkce jater (Child-Pughovo skóre > 9). (Viz body 4.4 a 5.2.)
Pacienti s poruchou funkce ledvin
U pacientů s lehkou poruchou funkce ledvin (odhadovaná rychlost glomerulární filtrace > 60 ml/min/1,73 m2) není vyžadována úprava dávkování. U pacientů s chronickým onemocněním ledvin a odhadovanou rychlostí glomerulární filtrace < 60 ml/min/1,73 m2 je doporučená dávka kombinace ezetimibu a simvastatinu 10/20 mg jednou denně večer (viz body 4.4, 5.1 a 5.2). Vyšší dávky je nutno podávat opatrně.
Způsob podání
Perorální podání. Přípravek Ezetimib/Simvastatin Mylan může být podáván jednou denně večer, s jídlem i bez jídla. Tableta se nesmí dělit.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku(y) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Těhotenství a kojení (viz bod 4.6).
Aktivní onemocnění jater nebo nevysvětlené přetrvávající zvýšení sérových aminotransferáz.
Současné podávání účinných inhibitorů CYP3A4 (látky, které zvyšují AUC přibližně pěti- nebo vícenásobně) (např. itrakonazolu, ketokonazolu, posakonazolu, vorikonazolu, erythromycinu, klarithromycinu, telithromycinu, inhibitorů HIV-proteázy (např. nelfinaviru), bocepreviru, telapreviru, nefazodonu a léčiv obsahujících kobicistat) (viz body 4.4 a 4.5).
Současné podávání gemfibrozilu, cyklosporinu nebo danazolu (viz body 4.4 a 4.5).
Současné podávání lomitapidu a ezetimibu/simvastatinu v dávce > 10/40 mg u pacientů s HoFH (viz body 4.2, 4.4 a 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Myopatie/Rhabdomyolýza
Po uvedení ezetimibu na trh byly popsány případy myopatie a rhabdomyolýzy. Většina pacientů, u nichž došlo k rozvoji rhabdomyolýzy, užívala statiny současně s ezetimibem. Rhabdomyolýza však byla velmi vzácně popsána při monoterapii ezetimibem a velmi vzácně po přidání ezetimibu k jiným látkám, o nichž je známo, že mohou vyvolat zvýšené riziko rozvoje rhabdomyolýzy.
Přípravek Ezetimib/Simvastatin Mylan obsahuje simvastatin. Simvastatin, podobně jako jiné inhibitory HMG-CoA reduktázy, ojediněle vyvolává myopatii projevující se svalovou bolestí, citlivostí nebo slabostí při hodnotách kreatinkinázy (CK) vyšších než 10násobek horní hranice normálu (upper limit of normal, ULN). Někdy má myopatie podobu rhabdomyolýzy bez akutního selhání ledvin nebo s ním na podkladě myoglobinurie a velmi vzácně se vyskytly případy úmrtí. Riziko myopatie se zvyšuje s vysokým stupněm aktivity inhibitorů HMG-CoA reduktázy v plazmě.
Podobně jako u jiných inhibitorů HMG-CoA reduktázy závisí riziko myopatie/rhabdomyolýzy na dávce simvastatinu. V databázi klinické studie, ve které bylo léčeno 41 413 pacientů simvastatinem, z nichž 24 747 (přibližně 60 %) bylo zařazeno do studií s mediánem doby pozorování alespoň 4 roky, byl výskyt myopatie přibližně 0,03 %, 0,08 % a 0,61 % při dávkách 20, 40 a 80 mg/den v uvedeném pořadí. V těchto studiích byli pacienti pečlivě sledováni a některé léčivé přípravky, u kterých dochází k interakci, byly vyloučeny.
V klinické studii, kde byli pacienti s infarktem myokardu v anamnéze léčeni simvastatinem v dávce 80 mg/den (střední hodnota doby pozorování 6,7 roku), byla incidence myopatie přibližně 1,0 % v porovnání s 0,02 % u pacientů léčených dávkou 20 mg/den. Přibližně polovina těchto případů myopatie se objevila během prvního roku léčby. Incidence myopatie během každého následujícího roku léčby byla přibližně 0,1 %. (Viz body 4.8 a 5.1.)
Riziko myopatie je v porovnání s jinými terapiemi založenými na statinech u pacientů léčených ezetimibem/simvastatinem v dávce 10/80 mg vyšší, přičemž účinnost ohledně snížení LDL-C je podobná. Proto se ezetimib/simvastatin v dávce 10/80 mg smí používat pouze u pacientů s těžkou hypercholesterolemií a s vysokým rizikem kardiovaskulárních komplikací, u kterých nebylo léčebných cílů dosaženo při nižších dávkách a tam, kde se předpokládá, že přínosy převáží nad potenciálními riziky. U pacientů užívajících ezetimib/simvastatin v dávce 10/80 mg, u nichž je potřebné interagující léčivo, se musí použít nižší dávka ezetimibu/simvastatinu nebo alternativní režim založený na statinech s nižším potenciálem k lékovým interakcím (viz dále Opatření ke snížení rizika myopatie způsobeného interakcemi léčivých přípravků a body 4.2, 4.3 a 4.5).
Ve studii IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) bylo randomizováno 18 144 pacientů s ischemickou chorobou srdeční a akutním koronárním syndromem v anamnéze do skupiny léčené ezetimibem/simvastatinem 10 mg/40 mg denně (n = 9 067) nebo 40 mg simvastatinu denně (n = 9 077). Během mediánu sledování 6,0 roku byla incidence myopatie u pacientů s ezetimibem/simvastatinem 0,2 % a u pacientů se simvastatinem 0,1 %, přičemž myopatie byla definována jako nevysvětlená svalová slabost nebo bolest s hladinou sérové kreatinkinázy (CK) > 10násobek ULN nebo s koncentrací CK > 5násobek a zároveň < 10násobek ULN ve dvou po sobě jdoucích měřeních. Incidence rhabdomyolýzy byla u pacientů s ezetimibem/simvastatinem 0,1 % a u pacientů se simvastatinem 0,2 %, přičemž rhabdomyolýza byla definována jako nevysvětlená svalová slabost nebo bolest s hladinou sérové CK > 10násobek ULN s prokázaným renálním poškozením, > 5násobek a zároveň < 10násobek ULN ve dvou po sobě jdoucích měřeních s prokázaným renálním poškozením nebo > 10 000 IU/l bez prokázaného renálního poškození (viz bod 4.8).
-
V klinické studii, ve které bylo randomizováno více než 9 000 pacientů s chronickým onemocněním ledvin do skupiny léčené ezetimibem/simvastatinem 10/20 mg denně (n = 4 650) nebo placebem (n = 4 620) (medián doby pozorování 4,9 roku), byla incidence myopatie 0,2 % u ezetimibu/simvastatinu a 0,1 % u placeba. (Viz bod 4.8.)
-
V klinické studii, ve které byli pacienti s vysokým rizikem kardiovaskulárního onemocnění léčeni simvastatinem v dávce 40 mg/den (medián doby sledování 3,9 roku), byla incidence myopatie přibližně 0,05 % u pacientů jiného než čínského původu (n = 7367) v porovnání s 0,24 % u pacientů čínského původu (n = 5468). Protože jediná asijská populace hodnocená v této klinické studii byla čínská, je třeba být opatrný při předepisování ezetimibu/simvastatinu asijským pacientům a musí být použita nejnižší možná dávka.
Snížení funkce transportních proteinů
Snížení funkce jaterních OATP transportních proteinů může zvyšovat systémovou expozici simvastatinu a zvyšovat riziko myopatie a rhabdomyolýzy. Snížení jejich funkce se může vyskytovat jako důsledek inhibice interagujícími přípravky (např. cyklosporin) nebo u pacientů, kteří jsou nosiči genotypu SLCO1B1 c.521T>C.
Pacienti, kteří jsou nosiči alely genu SLCO1B1 (c.521T>C) kódující méně aktivní OATP1B1 protein, mají zvýšenou systémovou expozici simvastatinu a zvýšené riziko myopatie. Riziko myopatie spojené s užíváním vysokých dávek simvastatinu (80 mg) je obecně, bez genetického testování, asi 1 %. Na základě výsledků studie SEARCH mají homozygotní nosiči alely C (nazýváni také CC), kteří jsou léčeni 80 mg simvastatinu, v průběhu jednoho roku léčby riziko vzniku myopatie 15 %, zatímco u heterozygotních nosičů alely C (CT) je riziko 1,5 %. U nosičů nejčastějšího genotypu (TT) (viz bod 5.2) je odpovídající riziko 0,3 %. Tam, kde je to možné, má být v rámci posouzení individuálních přínosů a rizik léčby před předepsáním simvastatinu v dávce 80 mg zváženo genetické vyšetření na přítomnost alely C a u nosičů genotypu CC se má od předepsání vysoké dávky upustit. Nepřítomnost tohoto genu při genetickém vyšetření ovšem nevylučuje, že se myopatie může vyskytnout.
Měření kreatinkinázy
Koncentrace kreatinkinázy (CK) se nesmí měřit po namáhavém fyzickém zatížení nebo při jakékoli jiné věrohodné příčině zvýšení CK, protože se tak ztěžuje interpretace hodnot. Pokud jsou koncentrace CK statisticky zvýšené při výchozím vyšetření (>5násobek ULN), je nutno k potvrzení výsledků stanovit koncentrace znovu za 5 až 7 dní.
Před léčbou
Všechny pacienty, u nichž se zahajuje léčba ezetimibem/simvastatinem nebo se dávka ezetimibu/simvastatinu zvyšuje, je nutno poučit o riziku myopatie a upozornit je na nutnost okamžitě ohlásit jakoukoli nevysvětlenou svalovou bolest, bolestivost nebo slabost.
Pozornost je třeba věnovat pacientům s faktory predispozice k rhabdomyolýze. Aby byla stanovena základní referenční hodnota, je nutno změřit koncentraci CK ještě před zahájením léčby v následujících případech:
- starší osoby (věk > 65 let)
- ženské pohlaví
- porucha funkce ledvin
- nekontrolovaná hypotyreóza
- dědičné onemocnění svalů v osobní nebo rodinné anamnéze
- svalová toxicita při použití statinu nebo fibrátu v anamnéze
- závislost na alkoholu (abúzus)
V takových případech je nutno zvážit riziko léčby v poměru k možnému přínosu a doporučuje se klinické sledování. Pokud již pacient dříve trpěl poruchou svalové funkce při užívání fibrátu nebo statinu, je nutno zahajovat léčbu jakýmkoli přípravkem obsahujícím statin (jako je přípravek Ezetimib/Simvastatin Mylan) s opatrností. Pokud jsou koncentrace CK při výchozím vyšetření statisticky významně zvýšeny (>5násobek ULN), nelze léčbu zahájit.
Během léčby
Jestliže během léčby přípravkem Ezetimib/Simvastatin Mylan pociťuje pacient svalovou bolest, slabost nebo křeče, je nutno mu změřit hodnotu CK. Pokud se zjistí, že hodnoty jsou i bez zvýšené tělesné námahy statisticky významně zvýšené (>5násobek ULN), je třeba léčbu ukončit. Pokud jsou svalové příznaky závažné a způsobují každodenní nepříjemné pocity, a to i pokud jsou hodnoty CK <5násobek ULN, je nutno zvážit vysazení léčby. Při podezření na myopatii z jiných příčin je nutno léčbu přerušit.
Výskyt imunitně zprostředkované nekrotizující myopatie (IMNM) v průběhu nebo po ukončení léčby některými statiny byl hlášen velmi vzácně. Klinicky je IMNM charakterizována perzistentní proximální svalovou slabostí a zvýšením sérové kreatinkinázy, která přetrvává navzdory přerušení léčby statiny.
Jestliže příznaky odezní a hodnota CK se vrátí k normálu, lze zvážit možnost obnovení léčby přípravkem Ezetimib/Simvastatin Mylan nebo jiným přípravkem obsahujícím statin v nejnižší dávce a při důsledném sledování pacienta.
Vyšší míra myopatie byla pozorována u pacientů titrovaných na dávku simvastatinu 80 mg (viz bod 5.1). Doporučuje se pravidelné měření kreatinkinázy, protože může být užitečné při identifikaci subklinických případů myopatie. Takové monitorování však nezaručuje, že se myopatii zabrání.
Léčbu kombinací ezetimibu a simvastatin je třeba dočasně vysadit několik dní před plánovaným chirurgickým zákrokem a pokud nastane vážný zdravotní nebo chirurgický stav.
Opatření ke snížení rizika myopatie způsobeného interakcemi léčivých přípravků (viz také bod 4.5)
Riziko myopatie a rhabdomyolýzy se významně zvyšuje současným podáváním ezetimibu/simvastatinu a účinných inhibitorů CYP3A4 (jako jsou itrakonazol, ketokonazol, posakonazol, vorikonazol, erythromycin, klarithromycin, telithromycin, inhibitory HIV-proteázy (např. nelfinavir), boceprevir, telaprevir, nefazodon, léčivé přípravky obsahující kobicistat), stejně jako cyklosporinu, danazolu a gemfibrozilu. Podávání těchto léčivých přípravků je kontraindikováno (viz bod 4.3).
Kvůli simvastatinu, složce přípravku Ezetimib/Simvastatin Mylan, se riziko myopatie a rhabdomyolýzy zvyšuje také při současném podávání dalších fibrátů, hypolipidemických dávek (>1 g/den) niacinu nebo při současném užívání amiodaronu, amlodipinu, verapamilu nebo diltiazemu s určitými dávkami ezetimibu/simvastatinu (viz body 4.2 a 4.5). Riziko myopatie včetně rhabdomyolýzy se může zvyšovat při současném podávání kyseliny fusidové s ezetimibem/simvastatinem. U pacientů s HoFH může být toto riziko zvýšeno užíváním lomitapidu současně s ezetimibem/simvastatinem. (viz bod 4.5).
Proto, pokud se týče inhibitorů CYP3A4, je současné užívání ezetimibu/simvastatinu s itrakonazolem, ketokonazolem, posakonazolem, vorikonazolem, inhibitory HIV-proteázy (např. nelfinavirem), boceprevirem, telaprevirem, erythromycinem, klarithromycinem, telithromycinem, nefazodonem a léčivými přípravky obsahujícími kobicistat kontraindikováno (viz body 4.3 a 4.5). Pokud je léčba silnými inhibitory CYP3A4 (látkami, které zvyšují AUC přibližně pěti- nebo vícenásobně) nezbytná, musí se po dobu léčby terapie ezetimibem/simvastatinem přerušit (a zvážit podávání jiného statinu). Navíc je nutná opatrnost při současném užívání ezetimibu/simvastatinu s některými dalšími méně účinnými inhibitory CYP3A4: flukonazolem, verapamilem, diltiazemem (viz body 4.2 a 4.5). Je nutno se vyvarovat současnému podávání ezetimibu/simvastatinu a požívání grapefruitové šťávy.
Simvastatin se nesmí podávat současně se systémovou léčbou kyselinou fusidovou nebo během 7 dnů po ukončení léčby kyselinou fusidovou. U pacientů, u kterých je systémová léčba kyselinou fusidovou považována za nezbytnou, se musí po dobu léčby kyselinou fusidovou přerušit léčba statinem. U pacientů léčených touto kombinací byly hlášeny případy rhabdomyolýzy (včetně fatálních) (viz bod 4.5). Pacienta je nutno poučit, aby ihned vyhledal lékařskou pomoc, pokud se objeví jakékoli příznaky slabosti, bolesti nebo citlivosti svalů.
Léčbu statiny lze znovu zahájit sedm dní po podání poslední dávky kyseliny fusidové. Za výjimečných okolností, kdy je potřebné dlouhodobé systémové podávání kyseliny fusidové, např. při závažných infekcích, je možno o potřebě současného podávání ezetimibu/simvastatinu a kyseliny fusidové uvažovat pouze případ od případu pod pečlivým lékařským dohledem.
Podávání ezetimibu/simvastatinu v dávkách nad 10/20 mg denně současně s hypolipidemickými dávkami niacinu (> 1 g/den) je třeba se vyhnout, pokud není pravděpodobné, že klinický přínos pro pacienta převáží nad zvýšeným rizikem myopatie (viz body 4.2 a 4.5.)
Současné podávání inhibitorů reduktázy HMG-CoA a niacinu (kyseliny nikotinové) v lipidy modifikujících dávkách ( 1g/den) bylo spojeno se vzácnými případy myopatie/rhabdomyolýzy, přičemž každá z těchto látek může myopatii způsobit sama.
V klinické studii (medián doby sledování 3,9 roku) zahrnující pacienty s vysokým rizikem kardiovaskulárního onemocnění a s dobře kontrolovanými hladinami LDL cholesterolu při dávce simvastatinu 40 mg/den s nebo bez ezetimibu v dávce 10 mg nebyl při přídavku lipidy modifikujících dávek (> 1 g/den) niacinu (kyseliny nikotinové) u kardiovaskulárních výsledků žádný přidaný benefit. Proto lékaři zvažující kombinovanou léčbu simvastatinem a niacinem (kyselinou nikotinovou) v lipidy modifikujících dávkách (>1 g/den) nebo přípravky s obsahem niacinu musí pečlivě zvážit možné přínosy a rizika a pacienty pečlivě sledovat s ohledem na jakékoli projevy a symptomy bolesti, citlivosti nebo slabosti svalů, zejména během prvních měsíců léčby a při zvyšování dávky kteréhokoli z léčivých přípravků.
Kromě toho byla v této studii incidence myopatie přibližně 0,24 % u pacientů čínského původu užívajících simvastatin v dávce 40 mg nebo kombinaci ezetimib/simvastatin v dávce 10/40 mg v porovnání s 1,24 % u pacientů čínského původu užívajících simvastatin v dávce 40 mg nebo kombinaci ezetimib/simvastatin v dávce 10/40 mg s kombinací kyselina nikotinová/laropiprant v dávce 2000 mg/40 mg s řízeným uvolňováním. Protože jediná asijská populace hodnocená v této studii byla čínská, a protože incidence myopatie je vyšší u pacientů čínského původu než u pacientů jiného původu, současné podávání kombinace ezetimibu a simvastatinu s lipidy modifikujícími dávkami (> 1 g/den) niacinu (kyseliny nikotinové) se u asijských pacientů nedoporučuje.
Acipimox je strukturně příbuzný s niacinem. Ačkoliv acipimox nebyl studován, riziko toxických účinků na svaly může být podobné jako u niacinu.
Kombinovaného užívání ezetimibu/simvastatinu v dávkách nad 10/20 mg denně současně s amiodaronem, amlodipinem, verapamilem nebo diltiazemem je nutno se vyvarovat. U pacientů s HoFH je třeba se vyvarovat současného užívání kombinace ezetimibu a simvastatinu v dávkách vyšších než 10/40 mg denně s lomitapidem. (Viz body 4.2, 4.3 a 4.5.)
Pacienti užívající spolu s kombinací ezetimibu a simvastatinu, zejména kombinace ezetimibu a simvastatinu obsahující vyšší dávky, jiné léčivé přípravky, u nichž je uvedeno, že v terapeutických dávkách mají středně silný inhibiční účinek na CYP3A4, mohou být vystaveni vyššímu riziku myopatie. Pokud se ezetimib/simvastatin podává současně se středně silnými inhibitory CYP3A4 (látky, které zvyšují AUC přibližně 2.5 násobně), může být nutná úprava dávky. U některých středně silných inhibitorů CYP3A4, např. diltiazemu, se doporučuje maximální dávka kombinace ezetimibu a simvastatinu 10/20 mg (viz bod 4.2).
Simvastatin je substrát efluxního transportéru BCRP (Breast Cancer Resistant Protein). Současné podávání přípravků, které jsou inhibitory BCRP (např. elbasvir a grazoprevir), může vést ke zvýšení plazmatické koncentrace simvastatinu a zvýšenému riziku myopatie; proto se má v závislosti na předepsané dávce zvážit úprava dávky simvastatinu. Současné podávání elbasviru a grazopreviru se simvastatinem nebylo studováno, nicméně u pacientů se současnou léčbou přípravky obsahujícími elbasvir nebo grazoprevir nemá dávka ezetimibu/simvastatinu přesáhnout 10/20 mg denně (viz bod 4.5).
Bezpečnost a účinnost kombinace ezetimibu a simvastatinu podávaného současně s fibráty nebyly sledovány. Existuje zvýšené riziko myopatie, jestliže je simvastatin užíván současně s fibráty (zejména s gemfibrozilem). Proto je současné užívání kombinace ezetimibu a simvastatinu s gemfibrozilem kontraindikováno (viz bod 4.3) a současné užívání s jinými fibráty se nedoporučuje (viz bod 4.5).
Jaterní enzymy
V kontrolovaných studiích současného podávání ezetimibu se simvastatinem bylo pozorováno postupné zvyšování koncentrací aminotransferáz. (> 3násobek ULN) (viz bod 4.8).
Ve studii IMPROVE-IT bylo randomizováno 18 144 pacientů s ischemickou chorobou srdeční a akutním koronárním syndromem v anamnéze do skupiny léčené ezetimibem/simvastatinem 10 mg/40 mg denně (n = 9 067) nebo 40 mg simvastatinu denně (n = 9 077). Během mediánu sledování 6,0 roku byla incidence následných zvýšení hladin aminotransferáz (> 3× ULN) 2,5 % u ezetimibu/simvastatinu a 2,3 % u simvastatinu (viz bod 4.8).
-
V klinické studii, ve které bylo randomizováno více než 9 000 pacientů s chronickým onemocněním ledvin do skupiny léčené kombinací ezetimibu a simvastatinu 10/20 mg denně (n = 4 650) nebo placebem (n = 4 620) (medián doby pozorování 4,9 roku), byla incidence následných zvýšení aminotransferáz (> 3 X ULN) 0,7 % u kombinace ezetimibu a simvastatinu a 0,6 % u placeba (viz bod 4.8).
Doporučuje se provést vyšetření jaterní funkce ještě před zahájením léčby kombinací ezetimibu a simvastatinu a dále kdykoli je to z klinického hlediska indikováno. U pacientů titrovaných na 10/80mg dávku je vždy nutno provést další vyšetření před titrací, 3 měsíce po titraci na 10/80mg dávku a dále pravidelně (např. každého půl roku) po dobu prvního roku léčby. Zvláštní pozornost je třeba věnovat pacientům se zvýšenými sérovými koncentracemi aminotransferáz a u těchto pacientů je nutno měření brzy opakovat a pak provádět častěji. Při zjištění progrese zvyšování koncentrací aminotransferáz, zvláště pokud se zvýší na trojnásobek ULN a zvýšení přetrvává, je nutno přípravek vysadit. Mějte na paměti, že ALT se může uvolňovat ze svalů, proto vzestup ALT s CK může ukazovat na myopatii (viz výše myopatie/rhabdomyolýza).
U pacientů užívajících statiny, včetně simvastatinu, existují vzácná poregistrační hlášení fatálního a nefatálního selhání jater. Pokud se během léčby kombinací ezetimibu a simvastatinu objeví závažné poškození jater s klinickými symptomy a/nebo hyperbilirubinemií nebo žloutenkou, léčbu je nutno ihned přerušit. Pokud se nezjistí žádná jiná etiologie, ezetimib/simvastatin znovu nenasazujte.
Ezetimib/simvastatin je nutno podávat opatrně pacientům konzumujícím alkohol ve větším množství.
Porucha funkce jater
Vzhledem k neznámým účinkům zvýšené expozice ezetimibu u pacientů se středně těžkou nebo těžkou poruchou funkce jater se nedoporučuje těmto jedincům ezetimib/simvastatin podávat (viz bod 5.2).
Diabetes mellitus
Některé důkazy naznačují, že statiny zvyšují hladinu glukózy v krvi a u některých pacientů s rizikem vzniku diabetu, mohou vyvolat hyperglykemii, která již vyžaduje diabetologickou péči. Toto riziko však nepřevažuje nad prospěchem léčby statiny – redukcí kardiovaskulárního rizika a není proto důvod pro ukončení léčby statiny. Pacienti se zvýšeným rizikem pro vznik diabetu (glukóza nalačno 5,6 až 6,9 mmol/l, BMI>30kg/m2, zvýšení triacylglycerolů v krvi, hypertenze) mají být klinicky a biochemicky monitorováni v souladu s národními doporučeními.
Pediatrická populace (10 až 17 let věku)
Účinnost a bezpečnost ezetimibu podávaného současně se simvastatinem pacientům ve věku 10 až 17 let s heterozygotní familiární hypercholesterolemií byla hodnocena v kontrolované klinické studii u dospívajících chlapců (Tannerův stupeň II nebo vyšší) a dívek, které již alespoň rok menstruovaly.
-
V této omezené kontrolované studii obecně nebyl u dospívajících chlapců nebo dívek prokázán žádný detekovatelný účinek na růst a pohlavní zrání, ani žádný účinek na menstruační cyklus u dívek. Nicméně účinky ezetimibu na růst a pohlavní zrání při trvání léčby > 33 týdnů nebyly hodnoceny (viz body 4.2 a 4.8).
Bezpečnost a účinnost ezetimibu podávaného současně s dávkami simvastatinu vyššími než 40 mg denně nebyly u pediatrických pacientů ve věku 10 až 17 let hodnoceny.
Ezetimib nebyl u pacientů mladších 10 let nebo dívek před první menstruací hodnocen. (Viz body 4.2 a 4.8.)
Dlouhodobá účinnost léčby ezetimibem u pacientů mladších 17 let s ohledem na snížení morbidity a mortality v dospělosti nebyla hodnocena.
Fibráty
Bezpečnost a účinnost ezetimibu podávaného společně s fibráty nebyly zkoumány (viz výše a body 4.3 a 4.5).
Antikoagulancia
Pokud se přidá ezetimib/simvastatin k warfarinu, jiným kumarinovým antikoagulanciím nebo fluindionu, je nutno řádně sledovat hodnotu International Normalised Ratio (INR) (viz bod 4.5).
Intersticiální plicní choroba
U některých statinů, včetně simvastatinu, byly hlášeny případy intersticiální plicní choroby, zvláště při dlouhodobé léčbě (viz bod 4.8). Příznaky mohou zahrnovat dušnost, neproduktivní kašel a celkové zhoršení zdravotního stavu (únava, úbytek tělesné hmotnosti a horečka). V případě podezření, že se u pacienta vyvinula intersticiální plicní choroba, musí být léčba ezetimibem/simvastatinem vysazena.
Pomocná látka
Tento léčivý přípravek obsahuje monohydrát laktosy. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, úplným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy nemají tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Farmakodynamické interakce
Interakce s hypolipidemiky, které mohou při samostatném podávání vyvolat myopatii
Riziko myopatie včetně rhabdomyolýzy se zvyšuje při současném podávání simvastatinu s fibráty. Navíc dochází k farmakokinetické interakci simvastatinu s gemfibrozilem, která vede ke zvýšení plazmatických koncentrací simvastatinu (viz dále Farmakokinetické interakce a body 4.3 a 4.4). Vzácné případy myopatie/rhabdomyolýzy se vyskytly při současném podávání simvastatinu s niacinem v lipidy modifikujících dávkách (> 1 g/den) (viz bod 4.4).
Fibráty mohou zvýšit vylučování cholesterolu do žluči s následným rozvojem cholelitiázy. V preklinické studii se psy zvýšil ezetimib cholesterol ve žlučníkové žluči (viz bod 5.3). I když význam tohoto preklinického zjištění pro člověka není znám, současné podávání kombinace ezetimibu a simvastatinu a fibrátů se nedoporučuje (viz bod 4.4).
Farmakokinetické interakce
Doporučení pro předepisování pro vzájemně reagující látky jsou shrnuty v tabulce níže (další podrobnosti jsou uvedeny v textu; viz také body 4.2, 4.3 a 4.4).
Lékové interakce související se zvýšeným rizikem myopatie/rhabdomyolýzy
| Reagující látky | Předepsaná doporučení |
| Silné inhibitory CYP3A4, např: Itrakonazol Ketokonazol Posakonazol Vorikonazol Erythromycin Klarithromycin Telithromycin Inhibitory HIV-proteázy (např. nelfinavir) Boceprevir Telaprevir Nefazodon Kobicistat Cyklosporin Danazol Gemfibrozil | Kontraindikováno s kombinací ezetimibu a simvastatinu |
| Jiné fibráty Kyselina fusidová | Nedoporučuje se podávat s kombinací ezetimibu a simvastatinu |
| Niacin (kyselina nikotinová) (> 1 g/den) | Nedoporučuje se podávat s kombinací ezetimibu a simvastatinu u asijských pacientů. |
| Amiodaron Amlodipin Verapamil Diltiazem Niacin (> 1 g/den) Elbasvir Grazoprevir | Nesmí být překročena dávka 10/20 mg kombinace ezetimibu a simvastatinu denně. |
| Lomitapid | U pacientů s HoFH nesmí být překročena dávka 10/40 mg kombinace ezetimibu a simvastatinu denně. |
| Grapefruitová šťáva | Je třeba se vyvarovat požití grapefruitové šťávy během podávání kombinace ezetimibu a simvastatinu |
Působení jiných léčivých přípravků na ezetimib/simvastatin
Kombinace ezetimibu a simvastatinu
Niacin: ve studii na 15 zdravých dospělých způsobilo souběžné podávání kombinace ezetimibu a simvastatinu (10/20 mg denně po dobu 7 dní) mírný vzestup středních hodnot plochy pod křivkou AUC niacinu (22 %) a nikotinylglycinu (19 %) podávaných v přípravku NIASPAN tablety s prodlouženým uvolňováním (1 000 mg po dobu 2 dní a 2 000 mg po dobu 5 dní po nízkotučné snídani). Ve stejné studii současně podávaný přípravek NIASPAN lehce zvyšoval střední hodnoty plochy pod křivkou AUC ezetimibu (9 %), celkového ezetimibu (26 %), simvastatinu (20 %) a kyseliny simvastatinové (35 %) (viz bod 4.2. a 4.4.).
Studie lékových interakcí s vyššími dávkami simvastatinu nebyly provedeny.
Ezetimib
Antacida: Současné podávání antacid snížilo rychlost absorpce ezetimibu, nemělo však žádný vliv na biologickou dostupnost ezetimibu. Tato snížená rychlost absorpce se nepovažuje za klinicky významnou.
Kolestyramin: Současné podávání kolestyraminu zmenšilo průměrnou velikost plochy pod křivkou (area under the curve, AUC) celkového ezetimibu (ezetimib + ezetimib glukuronid) přibližně o 55 %. Postupné snížení LDL-C při přidávání ezetimibu/simvastatinu k kolestyraminu se může touto interakcí zpomalit (viz bod 4.2).
Cyklosporin: Ve studii s osmi pacienty po transplantaci ledvin s clearance kreatininu >50 ml/min, kteří byli na stabilní dávce cyklosporinu, vedlo podávání jednotlivé 10 mg dávky ezetimibu ke 3,4násobnému (rozmezí 2,3–7,9násobné) zvýšení průměrné AUC celkového ezetimibu ve srovnání se zdravou populací z kontrolní skupiny, která dostávala ezetimib samostatně, z jiné studie (n = 17). V další studii se u pacienta po transplantaci ledvin s těžkou poruchou funkce ledvin, který dostával cyklosporin a další mnohonásobnou terapii, projevila 12násobně vyšší expozice celkovému ezetimibu ve srovnání se souběžnými kontrolními skupinami, které dostávaly ezetimib samostatně. V crossover studii ve dvou obdobích, která se provedla se 12 zdravými jedinci, vedlo denní podávání ezetimibu v dávce 20 mg po dobu 8 dní spolu s jednorázovým podáním cyklosporinu v dávce 100 mg sedmý den k průměrnému 15% zvětšení AUC cyklosporinu (rozmezí 10% pokles až 51% zvýšení) ve srovnání s jednorázovým podáním 100mg dávky samotného cyklosporinu. Kontrolovaná studie vlivu současného podávání ezetimibu na expozici cyklosporinu u pacientů po transplantaci ledvin nebyla dosud provedena. Současné podávání kombinace ezetimibu a simvastatinu a cyklosporinu je kontraindikováno (viz bod 4.3).
Fibráty: Současné podávání fenofibrátu nebo gemfibrozilu zvýšilo koncentrace celkového ezetimibu přibližně 1,5násobně a přibližně 1,7násobně v tomto pořadí. I když se uvedená zvýšení nepovažují za klinicky významná, současné podávání kombinace ezetimibu a simvastatinu s gemfibrozilem je kontraindikováno a s jinými fibráty se nedoporučuje (viz body 4.3 a 4.4).
Simvastatin
Simvastatin je substrátem cytochromu P450 3A4. Účinné inhibitory cytochromu P450 3A4 zvyšují riziko myopatie a rhabdomyolýzy tím, že zvyšují plazmatickou koncentraci inhibitorů HMG-CoA reduktázy během léčby simvastatinem. Mezi tyto inhibitory patří itrakonazol, ketokonazol, posakonazol, vorikonazol, erythromycin, klarithromycin, telithromycin, inhibitory HIV-proteázy (např. nelfinavir), boceprevir, telaprevir, nefazodon a léčivé přípravky obsahující kobicistat. Současné podávání itrakonazolu vedlo k více než 10násobnému zvýšení expozice kyselině simvastatinové (aktivní metabolit beta-hydroxykyseliny). Telithromycin vedl k 11násobnému zvýšení expozice kyselině simvastatinové.
Kombinace s itrakonazolem, ketokonazolem, posakonazolem, vorikonazolem, inhibitory HIV- proteázy (např. nelfinavirem), boceprevirem, telaprevirem, erythromycinem, klarithromycinem, telithromycinem, nefazodonem a léčivými přípravky obsahujícími kobicistat je kontraindikována, stejně tak s gemfibrozilem, cyklosporinem a danazolem (viz bod 4.3). Pokud je léčba silnými inhibitory CYP3A4 (látky, které zvyšují AUC přibližně pěti- nebo vícenásobně) nevyhnutelná, je nutno během této léčby podávání kombinace ezetimibu a simvastatinu dočasně přerušit (a zvážit podávání jiného statinu). Opatrnosti je třeba při podávání kombinace ezetimibu a simvastatinu s některými jinými, méně účinnými inhibitory CYP3A4: flukonazolem, verapamilem, diltiazemem (viz body 4.2 a 4.4).
Flukonazol: V souvislosti se současným podáváním simvastatinu a flukonazolu byly hlášeny vzácné případy rhabdomyolýzy. (viz bod 4.4).
Cyklosporin: Riziko rozvoje myopatie/rhabdomyolýzy se při současném podávání cyklosporinu a ezetimibu/simvastatinu zvyšuje. Proto je podávání spolu s cyklosporinem kontraindikováno (viz body 4.3 a 4.4). Přestože není mechanismus zcela objasněn, zdá se, že cyklosporin zvětšuje AUC inhibitorů HMG-CoA reduktázy. Zvětšení velikosti AUC kyseliny simvastatinové, je nejspíše zčásti v důsledku inhibice CYP3A4 a/nebo OATP1B1.
Danazol: Riziko rozvoje myopatie a rhabdomyolýzy se zvyšuje při současném podávání danazolu s ezetimibem/simvastatinem , proto je podávání danazolu kontraindikováno (viz body 4.3 a 4.4).
Gemfibrozil: Gemfibrozil zvětšuje velikost AUC simvastatinové kyseliny 1,9násobně, nejspíše v důsledku blokády dráhy glukuronidace a/nebo OATP1B1 (viz body 4.3 a 4.4). Současné podávání s gemfibrozilem je kontraindikováno.
Kyselina fusidová: Riziko myopatie včetně rhabdomyolýzy se při současném systémovém podávání kyseliny fusidové se statiny může zvyšovat. Mechanismus této interakce (zda jde o interakci farmakodynamickou nebo farmakokinetickou nebo obě) není dosud znám. U pacientů léčených touto kombinací byla hlášena rhabdomyolýza (včetně několika smrtelných případů). Současné podávání této kombinace může vést ke zvýšeným plazmatickým koncentracím obou látek. Pokud je léčba kyselinou fusidovou nezbytná, musí se po dobu léčby kyselinou fusidovou přerušit léčba ezetimibem/simvastatinem (viz bod 4.4.).
Amiodaron: Riziko myopatie a rhabdomyolýzy se zvyšuje při současném podávání amiodaronu se simvastatinem (viz bod 4.4). V klinické studii byla myopatie hlášena u 6 % pacientů léčených simvastatinem v dávce 80 mg a amiodaronem. Proto nesmí dávka ezetimibu/simvastatinu u pacientů současně léčených amiodaronem přesáhnout 10/20 mg denně.
Blokátory vápníkového kanálu
Verapamil
Riziko myopatie a rhabdomyolýzy je současným podáváním verapamilu se simvastatinem v dávce 40 nebo 80 mg zvýšeno (viz bod 4.4). Ve farmakokinetické studii vedlo současné podávání simvastatinu a verapamilu k 2,3násobnému zvýšení expozice kyselině simvastatinové, zčásti nejspíše v důsledku inhibice CYP3A4. Dávka ezetimibu/simvastatinu proto nesmí překročit hodnotu 10/20 mg denně u pacientů užívajících současně verapamil.
Diltiazem
Riziko myopatie a rhabdomyolýzy je současným podáváním diltiazemu se simvastatinem v dávce 80 mg zvýšeno (viz bod 4.4). Ve farmakokinetické studii vyvolalo současné podávání diltiazemu a simvastatinu 2,7násobné zvýšení expozice kyselině simvastatinové, pravděpodobně v důsledku inhibice CYP3A4. Dávka ezetimibu/simvastatinu proto nesmí překročit hodnotu 10/20 mg denně u pacientů současně užívajících diltiazem.
Amlodipin
Pacienti, kterým se podává amlodipin, léčení současně simvastatinem jsou vystaveni vyššímu riziku myopatie. Ve farmakokinetické studii způsobilo současné podání amlodipinu 1,6násobné zvýšení expozice kyselině simvastatinové. Proto u pacientů současně léčených amlodipinem nesmí dávka ezetimibu/simvastatinu přesáhnout 10/20 mg denně.
Lomitapid
Při současném podávání simvastatinu s lomitapidem může být riziko myopatie a rhabdomyolýzy zvýšeno (viz body 4.3 a 4.4). Proto u pacientů s HoFH, kteří užívají současně lomitapid, nesmí dávka ezetimibu/simvastatinu překročit 10/40 mg denně.
Středně silné inhibitory CYP3A4
Pacienti užívající spolu s kombinací ezetimibu a simvastatinu, zejména kombinace ezetimibu a simvastatinu obsahující vyšší dávky, jiná léčiva, u nichž je uvedeno, že mají středně silný inhibiční účinek na CYP3A4, mohou být vystaveni vyššímu riziku myopatie (viz bod 4.4).
Inhibitory transportního proteinu OATP1B1
Kyselina simvastatinová je substrát transportního proteinu OATP1B1. Současné podávání léčivých přípravků, které jsou inhibitory transportního proteinu OATP1B1, může vést ke zvýšeným plazmatickým koncentracím kyseliny simvastatinové a ke zvýšenému riziku myopatie (viz body 4.3 a 4.4).
Inhibitory BCRP (Breast Cancer Resistant Protein)
Současné podávání léčivých přípravků, které jsou inhibitory BCRP, včetně přípravků obsahujících elbasvir nebo grazoprevir, může vést ke zvýšení plazmatické koncentrace simvastatinu a zvýšenému riziku myopatie (viz body 4.2 a 4.4).
Grapefruitová šťáva
Grapefruitová šťáva inhibuje cytochrom P450 3A4. Současná konzumace velkých množství (nad 1 litr denně) grapefruitové šťávy a simvastatinu vedla k 7násobnému zvýšení expozice kyselině simvastatinové. Příjem 240 ml grapefruitové šťávy ráno a podání simvastatinu večer také vedlo k 1,9násobnému zvýšení. Konzumaci grapefruitové šťávy v průběhu léčby ezetimibem/simvastatinem je proto nutno se vyvarovat.
Kolchicin
Při současném podávání kolchicinu a simvastatinu se objevila hlášení myopatie a rhabdomyolýzy, a to u pacientů s poruchou funkce ledvin. U pacientů užívajících tuto kombinaci se doporučuje pečlivé klinické sledování.
Rifampicin
Jelikož je rifampicin silným induktorem CYP3A4, může se u pacientů dlouhodobě léčených rifampicinem (např. léčba tuberkulózy) objevit ztráta účinku simvastatinu. Ve farmakokinetické studii na normálních dobrovolnících byla při současném podání rifampicinu plocha pod křivkou průběhu koncentrace kyseliny simvastatinové v plazmě (AUC) snížena o 93 %.
Niacin
Byly pozorovány případy myopatie/rhabdomyolýzy při současném podávání simvastatinu a lipidy modifikujících dávek (> 1 g/den) niacinu (viz bod 4.4).
Účinky kombinace ezetimibu a simvastatinu na farmakokinetiku jiných léčivých přípravků
-
V preklinických studiích se ukázalo, že ezetimib neindukuje enzymy cytochromu P450 metabolizující léčivé látky. Nebyly pozorovány žádné klinicky významné farmakokinetické interakce mezi ezetimibem a léčivými látkami, o nichž je známo, že se metabolizují cytochromy P450 1A2, 2D6, 2C8, 2C9 a 3A4 nebo N-acetyltransferázou.
Antikoagulancia
Ve studii s 12 zdravými dospělými muži nemělo současné podávání ezetimibu (10 mg jednou denně) žádný statisticky významný vliv na biologickou dostupnost warfarinu a protrombinový čas.
-
V postmarketingových hlášeních se však objevily zprávy o zvýšených hodnotách International Normalised Ratio (INR) u pacientů, u nichž byl ezetimib přidán k warfarinu nebo fluindionu. V případech, kdy se ezetimib/simvastatin přidá k warfarinu, jinému kumarinovému antikoagulanciu nebo fluindionu, je nutno INR řádně sledovat (viz bod 4.4).
Simvastatin nemá na cytochrom P450 3A4 inhibiční účinek. Neočekává se tedy, že by simvastatin ovlivňoval plazmatické koncentrace látek biotransformovaných cytochromem P450 3A4.
Perorální antikoagulancia
Ve dvou klinických studiích, jedné se zdravými dobrovolníky a druhé s pacienty s hypercholesterolemií, simvastatin v dávkách 20–40 mg/denně mírně potencoval účinek kumarinových antikoagulancií: protrombinový čas, uváděný jako International Normalised Ratio (INR), se prodloužil z výchozí hodnoty 1,7 na 1,8 ve studii s dobrovolníky a z 2,6 na 3,4 ve studii s pacienty. Byly popsány velmi vzácné případy zvýšení INR. U pacientů užívajících kumarinová antikoagulancia je třeba před zahájením léčby ezetimibem/simvastatinem stanovit protrombinový čas a poté ho stanovovat dostatečně často v počátcích léčby, aby byla jistota, že nedochází k významným změnám protrombinového času. Po stabilizaci protrombinového času jej lze sledovat v intervalech obvykle doporučovaných pro pacienty užívající kumarinová antikoagulancia. Tento postup je třeba opakovat při změně dávky ezetimibu/simvastatinu nebo jeho vysazení. U pacientů, kteří neužívali antikoagulancia, nebyla léčba simvastatinem spojována s krvácením nebo se změnami protrombinového času.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Ateroskleróza je chronický děj a za normálních okolností by vysazení hypolipidemických léčivých přípravků během těhotenství mělo mít minimální dopad na dlouhodobé riziko související s primární hypercholesterolemií.
Kombinace ezetimibu a simvastatinu
Během těhotenství je kombinace ezetimibu a simvastatinu kontraindikována. O užívání kombinace ezetimibu a simvastatinu během těhotenství nejsou k dispozici žádné klinické údaje. Studie kombinační terapie u zvířat prokázaly reprodukční toxicitu. (Viz bod 5.3.)
Simvastatin
Bezpečnost simvastatinu u těhotných žen nebyla hodnocena. U těhotných žen se neprováděly žádné kontrolované klinické studie se simvastatinem. Vzácně se objevily zprávy o vrozených anomáliích po nitroděložní expozici inhibitorům HMG-CoA reduktázy. V analýze přibližně 200 prospektivně sledovaných těhotenství s expozicí simvastatinu nebo jinému blízce příbuzného inhibitoru HMG-CoA reduktázy v prvním trimestru byla incidence vrozených anomálií srovnatelná s hodnotou pozorovanou u celkové populace. Uvedený počet těhotenství byl ze statistického hlediska dostatečný k vyloučení 2,5násobného nebo většího nárůstu vrozených anomálií ve srovnání s běžnou incidencí.
Přestože nejsou k dispozici důkazy o tom, že by se výskyt vrozených anomálií u potomstva pacientů užívajících simvastatin nebo jiný blízce příbuzný inhibitor HMG-CoA reduktázy lišil od hodnot pozorovaných u celkové populace, může podávání simvastatinu nastávající matce snížit koncentrace mevalonátu u plodu; mevalonát je prekurzorem biologické syntézy cholesterolu. Z tohoto důvodu se nesmí kombinace ezetimibu a simvastatinu podávat těhotným ženám, ženám, které se snaží otěhotnět nebo ženám, které se domnívají, že jsou těhotné. Léčbu kombinací ezetimibu a simvastatinu je třeba vysadit na dobu těhotenství nebo do ověření, že žena není těhotná. (Viz bod 4.3.)
Ezetimib
O užívání ezetimibu během těhotenství nejsou k dispozici žádné klinické údaje.
Kojení
Během kojení je kombinace ezetimibu a simvastatinu kontraindikována. Studie s potkany prokázaly, že ezetimib se vylučuje do mateřského mléka. Není známo, zda se léčivé látky kombinace ezetimibu a simvastatinu vylučují do mateřského mléka. (Viz bod 4.3.)
Fertilita
Ezetimib
Nejsou k dispozici žádné údaje z klinických studií týkající se vlivu ezetimibu na fertilitu člověka. Ezetimib nemá žádný vliv na fertilitu samců nebo samic potkanů (viz bod 5.3).
Simvastatin
Nejsou k dispozici žádné údaje z klinických studií týkající se vlivu simvastatinu na fertilitu člověka. Simvastatin nemá žádný vliv na fertilitu samců nebo samic potkanů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit a používat stroje nebyly provedeny. Při řízení vozidel nebo ovládání strojů je však třeba vzít v úvahu, že byly hlášeny závratě.
4.8
Nežádoucí účinky
Tabulkový seznam nežádoucích účinků (klinické studie)
Ezetimib/simvastatin (nebo současné podávání ezetimibu a simvastatinu odpovídající kombinaci ezetimibu a simvastatinu) byl hodnocen z hlediska bezpečnosti v klinických studiích přibližně u 12 000 pacientů.
Častost výskytu nežádoucích účinků je seřazena následujícím způsobem: Velmi časté (>1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit) včetně izolovaných případů.
Následující nežádoucí účinky byly pozorovány u pacientů léčených kombinací ezetimibu a simvastatinu (n= 2 404) a s vyšší incidencí než u placeba (n= 1 340).
| Nežádoucí účinky kombinace ezetimibu a simvastatinu vyskytující se s vyšší incidencí než u placeba | ||
| Třída orgánových systémů | Časté | Méně časté |
| Psychiatrické poruchy | poruchy spánku | |
| Poruchy nervového systému | závrať; bolest hlavy | |
| Gastrointestinální poruchy | bolest břicha; břišní diskomfort; bolest v horní části břicha; dyspepsie; flatulence; nauzea; zvracení | |
| Poruchy kůže a podkožní tkáně | pruritus, vyrážka | |
| Poruchy svalové a kosterní soustavy a pojivové tkáně | artralgie; svalové spasmy; svalová slabost muskuloskeletální diskomfort; bolest krku; bolest končetin | |
| Celkové poruchy a reakce v místě aplikace | astenie; únava; malátnost; periferní edém | |
| Vyšetření | Zvýšení ALT a/nebo AST Vzestup kreatinkinázy v krvi | zvýšení bilirubinu v krvi, zvýšení kyseliny močové v krvi; zvýšení gama-glutamyltransferázy; zvýšení INR; přítomnost bílkoviny v moči; snížení tělesné hmotnosti |
Následující nežádoucí účinky byly pozorovány u pacientů léčených kombinací ezetimibu a simvastatinu (n = 9 595) a s vyšší incidencí než u statinů podávaných samotných (n = 8 883).
| Nežádoucí účinky kombinace ezetimibu a simvastatinu vyskytující se s vyšší incidencí než u statinů | ||
| Třída orgánových systémů | Časté | Méně časté |
| Psychiatrické poruchy | insomnie | |
| Poruchy nervového systému | bolest hlavy; parestezie | |
| Gastrointestinální poruchy | břišní distenze; průjem; sucho v ústech; dyspepsie; flatulence; refluxní choroba jícnu; zvracení | |
| Poruchy kůže a podkožní tkáně | pruritus; vyrážka; kopřivka | |
| Poruchy svalové a kosterní soustavy a pojivové tkáně | myalgie | artralgie; bolest zad; svalové spasmy; svalová slabost; muskuloskeletální bolest; bolest |
| končetin | ||
| Celkové poruchy a reakce v místě aplikace | astenie; bolest na hrudi; únava; periferní edém | |
| Vyšetření | zvýšení ALT a/nebo AST | zvýšení bilirubinu v krvi; zvýšení kreatinkinázy v krvi; zvýšení gammaglutamyltransferázy |
Pediatrická populace (od 10 do 17 let věku)
Ve studii zahrnující dospívající (10 až 17 let věku) pacienty s heterozygotní familiární hypercholesterolemií (n = 248) bylo u 3 % (4 pacienti) pacientů léčených kombinací ezetimibu a simvastatinu zjištěno zvýšení ALT a/nebo AST (> 3násobek horní hranice normálu, několikrát po sobě) v porovnání se 2 % (2 pacienti) ve skupině léčené simvastatinem v monoterapii; tato čísla byla ohledně zvýšení kreatinfosfokinázy (CPK) 2 % (2 pacienti), respektive 0 % (> 10násobek horní hranice normálu). Nebyly hlášeny žádné případy myopatie.
Hodnocení nebylo vhodné pro porovnání vzácných nežádoucích účinků.
Pacienti s ischemickou chorobou srdeční a akutním koronárním syndromem v anamnéze
Ve studii IMPROVE-IT (viz bod 5.1), která zahrnovala 18 144 pacientů léčených ezetimibem/simvastatinem 10 mg/40 mg (n = 9 067, z toho 6 % bylo titrováno na ezetimib/simvastatin 10 mg/80 mg) nebo simvastatinem v dávce 40 mg (n = 9 077, z toho 27 % bylo titrováno na 80 mg simvastatinu), byly bezpečnostní profily během mediánu sledování 6,0 roku podobné. Kvůli výskytu nežádoucích účinků ukončilo léčbu 10,6 % pacientů léčených ezetimibem/simvastatinem a 10,1 % pacientů léčených simvastatinem. Incidence myopatie byla u pacientů s ezetimibem/simvastatinem 0,2 % a u pacientů se simvastatinem 0,1 %, přičemž myopatie byla definována jako nevysvětlená svalová slabost nebo bolest s hladinou sérové CK > 10násobek ULN nebo s koncentrací CK > 5násobek a zároveň < 10násobek ULN ve dvou následných měřeních. Incidence rhabdomyolýzy byla u pacientů s ezetimibem/simvastatinem 0,1 % a u pacientů se simvastatinem 0,2 %, přičemž rhabdomyolýza byla definována jako nevysvětlená svalová slabost nebo bolest s hladinou sérové CK > 10násobek ULN s prokázaným renálním poškozením, > 5násobek a zároveň < 10násobek ULN ve dvou následných měřeních s prokázaným renálním poškozením nebo > 10 000 IU/l bez prokázaného renálního poškození. Incidence následných zvýšení hladin aminotransferáz (> 3× ULN) byla 2,5 % u ezetimibu/simvastatinu a 2,3 % u simvastatinu (viz bod 4.4.). Nežádoucí účinky související se žlučníkem byly hlášeny u 3,1 % pacientů léčených ezetimibem/simvastatinem a u 3,5 % pacientů léčených simvastatinem. Incidence hospitalizace kvůli cholecystektomii byla v obou skupinách 1,5 %. Rakovina (definovaná jako jakákoli nová malignita) byla během studie diagnostikována u 9,4 % pacientů léčených
ezetimibem/simvastatinem oproti 9,5 % pacientů léčených simvastatinem.
Pacienti s chronickým onemocněním ledvin
Ve studii „Study of Heart and Renal Protection“ (SHARP) (viz bod 5.1), která zahrnovala více než 9 000 pacientů léčených kombinací ezetimibu a simvastatinu 10/20 mg denně (n = 4 650) nebo placebem (n = 4 620), byly bezpečnostní profily srovnatelné v průběhu mediánu doby sledování 4,9 roku. V této studii byly zaznamenávány pouze závažné nežádoucí účinky a ukončení léčby kvůli případným nežádoucím účinkům. Míry ukončení léčby v důsledku nežádoucích účinků byly srovnatelné (10,4 % u pacientů léčených ezetimibem/simvastatinem, 9,8 % u pacientů léčených placebem). Incidence myopatie/rhabdomyolýzy byla 0,2 % u pacientů léčených ezetimibem/simvastatinem a 0,1 % u pacientů léčených placebem. Následné zvýšení aminotransferáz (> 3 X ULN) se vyskytlo u 0,7 % pacientů léčených ezetimibem/simvastatinem v porovnání s 0,6 % pacientů léčených placebem. V této studii nebyla žádná statisticky významná zvýšení incidence předem specifikovaných nežádoucích účinků, včetně rakoviny (9,4 % u ezetimibu/simvastatinu, 9,5 % u placeba), hepatitidy, cholycystektomie nebo komplikací žlučníkových kamenů či pankreatitidy.
Laboratorní hodnoty
Ve studiích současného podávání kombinace ezetimibu a simvastatinu s jinými přípravky dosáhla incidence klinicky významných zvýšení koncentrací sérových aminotransferáz (ALT a/nebo AST > 3násobek ULN, následující po sobě) u pacientů léčených ezetimibem/simvastatinem 1,7 %. Tato zvýšení byla obecně asymptomatická, nebyla spojena s rozvojem cholestázy a po vysazení terapie nebo při pokračování léčby se koncentrace vrátily na výchozí hodnoty (viz bod 4.4).
Klinicky významná zvýšení hodnot CK (> IQxULN) byla pozorována u 0,2 % pacientů užívajících ezetimib/simvastatin.
Zkušenosti po uvedení na trh
Následující nežádoucí účinky byly hlášeny během postmarketingového použití kombinace ezetimibu a simvastatinu nebo během klinických studií nebo během postmarketingovém použití každé z jednotlivých složek kombinace ezetimibu a simvastatinu.
| Není známo | |
| Poruchy krve a lymfatického systému |
|
| Poruchy metabolismu a výživy |
|
| Psychiatrické poruchy |
|
| Poruchy nervového systému |
|
| Cévní poruchy |
|
| Respirační, hrudní a mediastinální poruchy |
|
| Gastrointestinální poruchy |
|
| Poruchy jater a žlučových cest |
|
| Poruchy kůže a podkožní tkáně |
|
| Poruchy imunitního systému |
|
| Poruchy svalové a kosterní soustavy a pojivové tkáně |
|
| Poruchy reprodukčního systému a prsu |
|
| Celkové poruchy a reakce v místě aplikace |
|
* V klinické studii se myopatie vyskytovala často u pacientů léčených simvastatinem v dávce 80 mg/den v porovnání s pacienty léčenými dávkou 20 mg/den (1,0 % vs 0,02 %, v uvedeném pořadí) (viz body 4.4 a 4.5).
Velmi vzácně byla hlášena imunitně zprostředkovaná nekrotizující myopatie (IMNM), autoimunitní myopatie spojovaná s užíváním statinů. Pro IMNM je charakteristické: slabost proximálních svalových skupin a zvýšení kreatinkinázy v séru, které přetrvává navzdory vysazení léčby statiny; biopsie svalů ukazující nekrotizující myopatii bez významného zánětu; zlepšení po použití imunosupresivních látek (viz bod 4.4).
Zřejmý syndrom hypersenzitivity byl popsán vzácně, byly při něm pozorovány následující stavy: angioedém, lupusu podobný syndrom, revmatická polymyalgie, dermatomyozitida, vaskulitida, trombocytopenie, eozinofilie, zvýšená sedimentace erytrocytů, artritida a artralgie, kopřivka, fotosenzitivní reakce, pyrexie, zrudnutí, dyspnoe a malátnost.
Laboratorní hodnoty: zvýšení hodnoty alkalické fosfatázy, abnormální hodnoty jaterních testů.
U statinů, včetně simvastatinu, byla hlášena zvýšení hladin HbA1c a sérové glukózy nalačno.
V souvislosti s používáním statinů, včetně simvastatinu, byly zaznamenány vzácné poregistrační hlášení týkající se zhoršení kognitivních schopností (např. ztráta paměti, zapomnětlivost, amnézie, zhoršení paměti, zmatenost). Tato hlášení jsou obecně nezávažná a reverzibilní při vysazení statinů s proměnlivou dobou do nástupu (1 den až roky) a vymizení (medián 3 týdny) symptomů.
U některých statinů byly hlášeny následující nežádoucí účinky:
- Poruchy spánku, včetně nočních můr
- Sexuální dysfunkce
- Diabetes mellitus: Frekvence výskytu bude záviset na přítomnosti nebo absenci rizikových faktorů (glukóza nalačno > 5,6 mmol/l, BMI > 30 kg/m2, zvýšení triacylglycerolů v krvi, hypertenze v anamnéze).
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky: http:/
4.9 Předávkování
1. NÁZEV PŘÍPRAVKU
Ezetimib/Simvastatin Mylan 10 mg/10 mg tablety
Ezetimib/Simvastatin Mylan 10 mg/20 mg tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta 10 mg/10 mg obsahuje ezetimibum 10 mg a simvastatinum 10 mg.
Jedna tableta 10 mg/20 mg obsahuje ezetimibum 10 mg a simvastatinum 20 mg.
Pomocné látky se známým účinkem:
Jedna tableta 10 mg/10 mg obsahuje 68,85 mg monohydrátu laktózy.
Jedna tableta 10 mg/20 mg obsahuje 147,71 mg monohydrátu laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tableta.
Ezetimib/Simvastatin Mylan 10 mg/10 mg: bílé až téměř bílé, oválné, bikonvexní tablety velikosti přibližně 9 mm na délku a 4,5 mm na šířku s vyraženým M na jedné straně a ES1 na druhé straně.
Ezetimib/Simvastatin Mylan 10 mg/20 mg: bílé až téměř bílé, oválné, bikonvexní tablety velikosti přibližně 11 mm na délku a 6,5 mm na šířku s vyraženým M na jedné straně a ES2 na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence kardiovaskulárních příhod
Přípravek Ezetimib/Simvastatin Mylan je indikován ke snížení rizika kardiovaskulárních příhod (viz bod 5.1) u pacientů s ischemickou chorobou srdeční (ICHS) a s akutním koronárním syndromem (AKS) v anamnéze, bez ohledu na to, zda předtím byli léčeni statinem.
Hypercholesterolemie
Přípravek Ezetimib/Simvastatin Mylan je indikován jako přídatná terapie k dietě u pacientů s primární (heterozygotní familiární a nefamiliární) hypercholesterolemií nebo smíšenou hyperlipidemií, kde je vhodné použití kombinovaného přípravku:
- u pacientů, kteří nejsou samotným statinem dostatečně kontrolováni
- u pacientů, kteří se již léčí statinem a ezetimibem.
Homozygotní familiární hypercholesterolemie (HoFH)
Přípravek Ezetimib/Simvastatin Mylan je indikován jako přídatná terapie k dietě u pacientů s HoFH. Pacienti mohou dostávat i další přídatnou terapii (např. aferézu nízkodenzitního lipoproteinu [LDL]).
4.2 Dávkování a způsob podání
Text odkazuje i na síly přípravku 10 mg/40 mg a 10 mg/80 mg. Tyto síly nejsou ovšem v České republice pod názvem Ezetimib/Simvastatin Mylan registrovány. Může se ale stát, že budete tyto síly užívat v podobě přípravku od jiného držitele rozhodnutí o registraci, proto i těmto informacím věnujte pozornost.
Dávkování
Hypercholesterolemie
Pacient má být na odpovídající hypolipidemické dietě a v průběhu léčby přípravkem Ezetimib/Simvastatin Mylan má v dietě pokračovat.
Dávkovací rozmezí kombinace ezetimibu a simvastatinu je od 10/10 mg/den až po 10/80 mg/den večer. Všechny velikosti dávky nemusí být k dispozici ve všech členských státech. Běžná dávka je 10/20 mg/den nebo 10/40 mg/den podána jednorázově večer. 10/80mg dávka se doporučuje pouze u pacientů se závažnou hypercholesterolemií a s vysokým rizikem kardiovaskulárních komplikací, u kterých se při nižších dávkách nedosáhlo léčebných cílů a pokud se očekává, že přínosy převáží nad potenciálními riziky (viz body 4.4 a 5.1). Při zahajování léčby nebo úpravě dávky je třeba vzít v úvahu koncentraci cholesterolu nízkodenzitního lipoproteinu (low-density lipoprotein cholesterol, LDL-C), riziko ischemické choroby srdeční a odpověď na aktuální cholesterol snižující léčbu.
Dávku přípravku Ezetimib/Simvastatin Mylan je nutno individuálně upravit podle známé účinnosti různých sil dávek kombinace ezetimibu a simvastatinu (viz bod 5.1, Tabulka 1) a podle odpovědi na aktuální cholesterol snižující léčbu. Úprava dávky, pokud je zapotřebí, se musí provádět s odstupem minimálně 4 týdnů.
Pacienti s ischemickou chorobou srdeční a akutním koronárním syndromem v anamnéze
Ve studii zaměřené na snížení rizika kardiovaskulárních příhod (IMPROVE-IT) byla počáteční dávka 10/40 mg jednou denně večer. Dávka 10/80 mg se doporučuje pouze za předpokladu, že přínosy převáží nad potenciálními riziky.
Homozygotní familiární hypercholesterolemie
Doporučená dávka kombinace ezetimibu a simvastatinu pro pacienty s homozygotní familiární hypercholesterolemií je 10/40 mg/den večer. Dávka 10/80 mg se doporučuje, pouze pokud se předpokládá, že přínosy převáží nad potenciálními riziky (viz dále; body 4.3 a 4.4).
Přípravek Ezetimib/Simvastatin Mylan lze použít jako přídatný přípravek k dalším formám hypolipidemické léčby (např. LDL aferéza) u těchto pacientů, nebo pokud není taková léčba k dispozici.
U pacientů užívajících lomitapid současně s přípravkem Ezetimib/Simvastatin Mylan nesmí dávka kombinace ezetimibu a simvastatinu překročit množství 10/40 mg denně (viz body 4.3, 4.4 a 4.5).
Současné podávání s jinými léčivými přípravky
Přípravek Ezetimib/Simvastatin Mylan je nutno podávat > 2 hodiny před nebo > 4 hodiny po podání sekvestrantu žlučových kyselin.
U pacientů užívajících amiodaron, amlodipin, verapamil, diltiazem nebo přípravky obsahující elbasvir nebo grazoprevir současně s kombinací ezetimibu a simvastatinu nesmí dávka přípravku Ezetimib/Simvastatin Mylan překročit hodnotu 10/20 mg/den (viz body 4.4 a 4.5).
U pacientů, kteří užívají hypolipidemické dávky niacinu (> 1 g/denně) současně s kombinací ezetimibu a simvastatinu, nesmí dávka ezetimibu/simvastatinu překročit hodnotu 10/20 mg/den (viz body 4.4 a 4.5).
Starší pacienti
U starších pacientů není nutno dávku upravovat (viz bod 5.2).
Pediatrická populace
Léčba musí být zahájena pod dohledem specialisty.
Dospívající > 10 let věku (pubertální status: chlapci Tannerův stupeň II a vyšší a dívky, které již alespoň jeden rok menstruují): klinické zkušenosti u pediatrických a dospívajících pacientů (ve věku 10 až 17 let) jsou omezené. Obvyklá doporučená zahajovací dávka je 10/10 mg jednou denně večer. Doporučené dávkové rozmezí je 10/10 až maximálně 10/40 mg/den (viz body 4.4 a 5.2).
Děti < 10 let: kombinace ezetimibu a simvastatinu se u dětí mladších 10 let kvůli nedostatku údajů o bezpečnosti a účinnosti nedoporučuje (viz bod 5.2). Zkušenosti s dětmi před pubertou jsou omezené.
Pacienti s poruchou funkce jater
U pacientů s lehkou poruchou funkce jater (Child-Pughovo skóre 5 až 6) není nutno dávkování upravovat. Nedoporučuje se podávat kombinaci ezetimibu a simvastatinu pacientům se středně těžkou (Child-Pughovo skóre 7 až 9) nebo těžkou poruchou funkce jater (Child-Pughovo skóre > 9). (Viz body 4.4 a 5.2.)
Pacienti s poruchou funkce ledvin
U pacientů s lehkou poruchou funkce ledvin (odhadovaná rychlost glomerulární filtrace > 60 ml/min/1,73 m2) není vyžadována úprava dávkování. U pacientů s chronickým onemocněním ledvin a odhadovanou rychlostí glomerulární filtrace < 60 ml/min/1,73 m2 je doporučená dávka kombinace ezetimibu a simvastatinu 10/20 mg jednou denně večer (viz body 4.4, 5.1 a 5.2). Vyšší dávky je nutno podávat opatrně.
Způsob podání
Perorální podání. Přípravek Ezetimib/Simvastatin Mylan může být podáván jednou denně večer, s jídlem i bez jídla. Tableta se nesmí dělit.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku(y) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Těhotenství a kojení (viz bod 4.6).
Aktivní onemocnění jater nebo nevysvětlené přetrvávající zvýšení sérových aminotransferáz.
Současné podávání účinných inhibitorů CYP3A4 (látky, které zvyšují AUC přibližně pěti- nebo vícenásobně) (např. itrakonazolu, ketokonazolu, posakonazolu, vorikonazolu, erythromycinu, klarithromycinu, telithromycinu, inhibitorů HIV-proteázy (např. nelfinaviru), bocepreviru, telapreviru, nefazodonu a léčiv obsahujících kobicistat) (viz body 4.4 a 4.5).
Současné podávání gemfibrozilu, cyklosporinu nebo danazolu (viz body 4.4 a 4.5).
Současné podávání lomitapidu a ezetimibu/simvastatinu v dávce > 10/40 mg u pacientů s HoFH (viz body 4.2, 4.4 a 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Myopatie/Rhabdomyolýza
Po uvedení ezetimibu na trh byly popsány případy myopatie a rhabdomyolýzy. Většina pacientů, u nichž došlo k rozvoji rhabdomyolýzy, užívala statiny současně s ezetimibem. Rhabdomyolýza však byla velmi vzácně popsána při monoterapii ezetimibem a velmi vzácně po přidání ezetimibu k jiným látkám, o nichž je známo, že mohou vyvolat zvýšené riziko rozvoje rhabdomyolýzy.
Přípravek Ezetimib/Simvastatin Mylan obsahuje simvastatin. Simvastatin, podobně jako jiné inhibitory HMG-CoA reduktázy, ojediněle vyvolává myopatii projevující se svalovou bolestí, citlivostí nebo slabostí při hodnotách kreatinkinázy (CK) vyšších než 10násobek horní hranice normálu (upper limit of normal, ULN). Někdy má myopatie podobu rhabdomyolýzy bez akutního selhání ledvin nebo s ním na podkladě myoglobinurie a velmi vzácně se vyskytly případy úmrtí. Riziko myopatie se zvyšuje s vysokým stupněm aktivity inhibitorů HMG-CoA reduktázy v plazmě.
Podobně jako u jiných inhibitorů HMG-CoA reduktázy závisí riziko myopatie/rhabdomyolýzy na dávce simvastatinu. V databázi klinické studie, ve které bylo léčeno 41 413 pacientů simvastatinem, z nichž 24 747 (přibližně 60 %) bylo zařazeno do studií s mediánem doby pozorování alespoň 4 roky, byl výskyt myopatie přibližně 0,03 %, 0,08 % a 0,61 % při dávkách 20, 40 a 80 mg/den v uvedeném pořadí. V těchto studiích byli pacienti pečlivě sledováni a některé léčivé přípravky, u kterých dochází k interakci, byly vyloučeny.
V klinické studii, kde byli pacienti s infarktem myokardu v anamnéze léčeni simvastatinem v dávce 80 mg/den (střední hodnota doby pozorování 6,7 roku), byla incidence myopatie přibližně 1,0 % v porovnání s 0,02 % u pacientů léčených dávkou 20 mg/den. Přibližně polovina těchto případů myopatie se objevila během prvního roku léčby. Incidence myopatie během každého následujícího roku léčby byla přibližně 0,1 %. (Viz body 4.8 a 5.1.)
Riziko myopatie je v porovnání s jinými terapiemi založenými na statinech u pacientů léčených ezetimibem/simvastatinem v dávce 10/80 mg vyšší, přičemž účinnost ohledně snížení LDL-C je podobná. Proto se ezetimib/simvastatin v dávce 10/80 mg smí používat pouze u pacientů s těžkou hypercholesterolemií a s vysokým rizikem kardiovaskulárních komplikací, u kterých nebylo léčebných cílů dosaženo při nižších dávkách a tam, kde se předpokládá, že přínosy převáží nad potenciálními riziky. U pacientů užívajících ezetimib/simvastatin v dávce 10/80 mg, u nichž je potřebné interagující léčivo, se musí použít nižší dávka ezetimibu/simvastatinu nebo alternativní režim založený na statinech s nižším potenciálem k lékovým interakcím (viz dále Opatření ke snížení rizika myopatie způsobeného interakcemi léčivých přípravků a body 4.2, 4.3 a 4.5).
Ve studii IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) bylo randomizováno 18 144 pacientů s ischemickou chorobou srdeční a akutním koronárním syndromem v anamnéze do skupiny léčené ezetimibem/simvastatinem 10 mg/40 mg denně (n = 9 067) nebo 40 mg simvastatinu denně (n = 9 077). Během mediánu sledování 6,0 roku byla incidence myopatie u pacientů s ezetimibem/simvastatinem 0,2 % a u pacientů se simvastatinem 0,1 %, přičemž myopatie byla definována jako nevysvětlená svalová slabost nebo bolest s hladinou sérové kreatinkinázy (CK) > 10násobek ULN nebo s koncentrací CK > 5násobek a zároveň < 10násobek ULN ve dvou po sobě jdoucích měřeních. Incidence rhabdomyolýzy byla u pacientů s ezetimibem/simvastatinem 0,1 % a u pacientů se simvastatinem 0,2 %, přičemž rhabdomyolýza byla definována jako nevysvětlená svalová slabost nebo bolest s hladinou sérové CK > 10násobek ULN s prokázaným renálním poškozením, > 5násobek a zároveň < 10násobek ULN ve dvou po sobě jdoucích měřeních s prokázaným renálním poškozením nebo > 10 000 IU/l bez prokázaného renálního poškození (viz bod 4.8).
-
V klinické studii, ve které bylo randomizováno více než 9 000 pacientů s chronickým onemocněním ledvin do skupiny léčené ezetimibem/simvastatinem 10/20 mg denně (n = 4 650) nebo placebem (n = 4 620) (medián doby pozorování 4,9 roku), byla incidence myopatie 0,2 % u ezetimibu/simvastatinu a 0,1 % u placeba. (Viz bod 4.8.)
-
V klinické studii, ve které byli pacienti s vysokým rizikem kardiovaskulárního onemocnění léčeni simvastatinem v dávce 40 mg/den (medián doby sledování 3,9 roku), byla incidence myopatie přibližně 0,05 % u pacientů jiného než čínského původu (n = 7367) v porovnání s 0,24 % u pacientů čínského původu (n = 5468). Protože jediná asijská populace hodnocená v této klinické studii byla čínská, je třeba být opatrný při předepisování ezetimibu/simvastatinu asijským pacientům a musí být použita nejnižší možná dávka.
Snížení funkce transportních proteinů
Snížení funkce jaterních OATP transportních proteinů může zvyšovat systémovou expozici simvastatinu a zvyšovat riziko myopatie a rhabdomyolýzy. Snížení jejich funkce se může vyskytovat jako důsledek inhibice interagujícími přípravky (např. cyklosporin) nebo u pacientů, kteří jsou nosiči genotypu SLCO1B1 c.521T>C.
Pacienti, kteří jsou nosiči alely genu SLCO1B1 (c.521T>C) kódující méně aktivní OATP1B1 protein, mají zvýšenou systémovou expozici simvastatinu a zvýšené riziko myopatie. Riziko myopatie spojené s užíváním vysokých dávek simvastatinu (80 mg) je obecně, bez genetického testování, asi 1 %. Na základě výsledků studie SEARCH mají homozygotní nosiči alely C (nazýváni také CC), kteří jsou léčeni 80 mg simvastatinu, v průběhu jednoho roku léčby riziko vzniku myopatie 15 %, zatímco u heterozygotních nosičů alely C (CT) je riziko 1,5 %. U nosičů nejčastějšího genotypu (TT) (viz bod 5.2) je odpovídající riziko 0,3 %. Tam, kde je to možné, má být v rámci posouzení individuálních přínosů a rizik léčby před předepsáním simvastatinu v dávce 80 mg zváženo genetické vyšetření na přítomnost alely C a u nosičů genotypu CC se má od předepsání vysoké dávky upustit. Nepřítomnost tohoto genu při genetickém vyšetření ovšem nevylučuje, že se myopatie může vyskytnout.
Měření kreatinkinázy
Koncentrace kreatinkinázy (CK) se nesmí měřit po namáhavém fyzickém zatížení nebo při jakékoli jiné věrohodné příčině zvýšení CK, protože se tak ztěžuje interpretace hodnot. Pokud jsou koncentrace CK statisticky zvýšené při výchozím vyšetření (>5násobek ULN), je nutno k potvrzení výsledků stanovit koncentrace znovu za 5 až 7 dní.
Před léčbou
Všechny pacienty, u nichž se zahajuje léčba ezetimibem/simvastatinem nebo se dávka ezetimibu/simvastatinu zvyšuje, je nutno poučit o riziku myopatie a upozornit je na nutnost okamžitě ohlásit jakoukoli nevysvětlenou svalovou bolest, bolestivost nebo slabost.
Pozornost je třeba věnovat pacientům s faktory predispozice k rhabdomyolýze. Aby byla stanovena základní referenční hodnota, je nutno změřit koncentraci CK ještě před zahájením léčby v následujících případech:
- starší osoby (věk > 65 let)
- ženské pohlaví
- porucha funkce ledvin
- nekontrolovaná hypotyreóza
- dědičné onemocnění svalů v osobní nebo rodinné anamnéze
- svalová toxicita při použití statinu nebo fibrátu v anamnéze
- závislost na alkoholu (abúzus)
V takových případech je nutno zvážit riziko léčby v poměru k možnému přínosu a doporučuje se klinické sledování. Pokud již pacient dříve trpěl poruchou svalové funkce při užívání fibrátu nebo statinu, je nutno zahajovat léčbu jakýmkoli přípravkem obsahujícím statin (jako je přípravek Ezetimib/Simvastatin Mylan) s opatrností. Pokud jsou koncentrace CK při výchozím vyšetření statisticky významně zvýšeny (>5násobek ULN), nelze léčbu zahájit.
Během léčby
Jestliže během léčby přípravkem Ezetimib/Simvastatin Mylan pociťuje pacient svalovou bolest, slabost nebo křeče, je nutno mu změřit hodnotu CK. Pokud se zjistí, že hodnoty jsou i bez zvýšené tělesné námahy statisticky významně zvýšené (>5násobek ULN), je třeba léčbu ukončit. Pokud jsou svalové příznaky závažné a způsobují každodenní nepříjemné pocity, a to i pokud jsou hodnoty CK <5násobek ULN, je nutno zvážit vysazení léčby. Při podezření na myopatii z jiných příčin je nutno léčbu přerušit.
Výskyt imunitně zprostředkované nekrotizující myopatie (IMNM) v průběhu nebo po ukončení léčby některými statiny byl hlášen velmi vzácně. Klinicky je IMNM charakterizována perzistentní proximální svalovou slabostí a zvýšením sérové kreatinkinázy, která přetrvává navzdory přerušení léčby statiny.
Jestliže příznaky odezní a hodnota CK se vrátí k normálu, lze zvážit možnost obnovení léčby přípravkem Ezetimib/Simvastatin Mylan nebo jiným přípravkem obsahujícím statin v nejnižší dávce a při důsledném sledování pacienta.
Vyšší míra myopatie byla pozorována u pacientů titrovaných na dávku simvastatinu 80 mg (viz bod 5.1). Doporučuje se pravidelné měření kreatinkinázy, protože může být užitečné při identifikaci subklinických případů myopatie. Takové monitorování však nezaručuje, že se myopatii zabrání.
Léčbu kombinací ezetimibu a simvastatin je třeba dočasně vysadit několik dní před plánovaným chirurgickým zákrokem a pokud nastane vážný zdravotní nebo chirurgický stav.
Opatření ke snížení rizika myopatie způsobeného interakcemi léčivých přípravků (viz také bod 4.5)
Riziko myopatie a rhabdomyolýzy se významně zvyšuje současným podáváním ezetimibu/simvastatinu a účinných inhibitorů CYP3A4 (jako jsou itrakonazol, ketokonazol, posakonazol, vorikonazol, erythromycin, klarithromycin, telithromycin, inhibitory HIV-proteázy (např. nelfinavir), boceprevir, telaprevir, nefazodon, léčivé přípravky obsahující kobicistat), stejně jako cyklosporinu, danazolu a gemfibrozilu. Podávání těchto léčivých přípravků je kontraindikováno (viz bod 4.3).
Kvůli simvastatinu, složce přípravku Ezetimib/Simvastatin Mylan, se riziko myopatie a rhabdomyolýzy zvyšuje také při současném podávání dalších fibrátů, hypolipidemických dávek (>1 g/den) niacinu nebo při současném užívání amiodaronu, amlodipinu, verapamilu nebo diltiazemu s určitými dávkami ezetimibu/simvastatinu (viz body 4.2 a 4.5). Riziko myopatie včetně rhabdomyolýzy se může zvyšovat při současném podávání kyseliny fusidové s ezetimibem/simvastatinem. U pacientů s HoFH může být toto riziko zvýšeno užíváním lomitapidu současně s ezetimibem/simvastatinem. (viz bod 4.5).
Proto, pokud se týče inhibitorů CYP3A4, je současné užívání ezetimibu/simvastatinu s itrakonazolem, ketokonazolem, posakonazolem, vorikonazolem, inhibitory HIV-proteázy (např. nelfinavirem), boceprevirem, telaprevirem, erythromycinem, klarithromycinem, telithromycinem, nefazodonem a léčivými přípravky obsahujícími kobicistat kontraindikováno (viz body 4.3 a 4.5). Pokud je léčba silnými inhibitory CYP3A4 (látkami, které zvyšují AUC přibližně pěti- nebo vícenásobně) nezbytná, musí se po dobu léčby terapie ezetimibem/simvastatinem přerušit (a zvážit podávání jiného statinu). Navíc je nutná opatrnost při současném užívání ezetimibu/simvastatinu s některými dalšími méně účinnými inhibitory CYP3A4: flukonazolem, verapamilem, diltiazemem (viz body 4.2 a 4.5). Je nutno se vyvarovat současnému podávání ezetimibu/simvastatinu a požívání grapefruitové šťávy.
Simvastatin se nesmí podávat současně se systémovou léčbou kyselinou fusidovou nebo během 7 dnů po ukončení léčby kyselinou fusidovou. U pacientů, u kterých je systémová léčba kyselinou fusidovou považována za nezbytnou, se musí po dobu léčby kyselinou fusidovou přerušit léčba statinem. U pacientů léčených touto kombinací byly hlášeny případy rhabdomyolýzy (včetně fatálních) (viz bod 4.5). Pacienta je nutno poučit, aby ihned vyhledal lékařskou pomoc, pokud se objeví jakékoli příznaky slabosti, bolesti nebo citlivosti svalů.
Léčbu statiny lze znovu zahájit sedm dní po podání poslední dávky kyseliny fusidové. Za výjimečných okolností, kdy je potřebné dlouhodobé systémové podávání kyseliny fusidové, např. při závažných infekcích, je možno o potřebě současného podávání ezetimibu/simvastatinu a kyseliny fusidové uvažovat pouze případ od případu pod pečlivým lékařským dohledem.
Podávání ezetimibu/simvastatinu v dávkách nad 10/20 mg denně současně s hypolipidemickými dávkami niacinu (> 1 g/den) je třeba se vyhnout, pokud není pravděpodobné, že klinický přínos pro pacienta převáží nad zvýšeným rizikem myopatie (viz body 4.2 a 4.5.)
Současné podávání inhibitorů reduktázy HMG-CoA a niacinu (kyseliny nikotinové) v lipidy modifikujících dávkách ( 1g/den) bylo spojeno se vzácnými případy myopatie/rhabdomyolýzy, přičemž každá z těchto látek může myopatii způsobit sama.
V klinické studii (medián doby sledování 3,9 roku) zahrnující pacienty s vysokým rizikem kardiovaskulárního onemocnění a s dobře kontrolovanými hladinami LDL cholesterolu při dávce simvastatinu 40 mg/den s nebo bez ezetimibu v dávce 10 mg nebyl při přídavku lipidy modifikujících dávek (> 1 g/den) niacinu (kyseliny nikotinové) u kardiovaskulárních výsledků žádný přidaný benefit. Proto lékaři zvažující kombinovanou léčbu simvastatinem a niacinem (kyselinou nikotinovou) v lipidy modifikujících dávkách (>1 g/den) nebo přípravky s obsahem niacinu musí pečlivě zvážit možné přínosy a rizika a pacienty pečlivě sledovat s ohledem na jakékoli projevy a symptomy bolesti, citlivosti nebo slabosti svalů, zejména během prvních měsíců léčby a při zvyšování dávky kteréhokoli z léčivých přípravků.
Kromě toho byla v této studii incidence myopatie přibližně 0,24 % u pacientů čínského původu užívajících simvastatin v dávce 40 mg nebo kombinaci ezetimib/simvastatin v dávce 10/40 mg v porovnání s 1,24 % u pacientů čínského původu užívajících simvastatin v dávce 40 mg nebo kombinaci ezetimib/simvastatin v dávce 10/40 mg s kombinací kyselina nikotinová/laropiprant v dávce 2000 mg/40 mg s řízeným uvolňováním. Protože jediná asijská populace hodnocená v této studii byla čínská, a protože incidence myopatie je vyšší u pacientů čínského původu než u pacientů jiného původu, současné podávání kombinace ezetimibu a simvastatinu s lipidy modifikujícími dávkami (> 1 g/den) niacinu (kyseliny nikotinové) se u asijských pacientů nedoporučuje.
Acipimox je strukturně příbuzný s niacinem. Ačkoliv acipimox nebyl studován, riziko toxických účinků na svaly může být podobné jako u niacinu.
Kombinovaného užívání ezetimibu/simvastatinu v dávkách nad 10/20 mg denně současně s amiodaronem, amlodipinem, verapamilem nebo diltiazemem je nutno se vyvarovat. U pacientů s HoFH je třeba se vyvarovat současného užívání kombinace ezetimibu a simvastatinu v dávkách vyšších než 10/40 mg denně s lomitapidem. (Viz body 4.2, 4.3 a 4.5.)
Pacienti užívající spolu s kombinací ezetimibu a simvastatinu, zejména kombinace ezetimibu a simvastatinu obsahující vyšší dávky, jiné léčivé přípravky, u nichž je uvedeno, že v terapeutických dávkách mají středně silný inhibiční účinek na CYP3A4, mohou být vystaveni vyššímu riziku myopatie. Pokud se ezetimib/simvastatin podává současně se středně silnými inhibitory CYP3A4 (látky, které zvyšují AUC přibližně 2.5 násobně), může být nutná úprava dávky. U některých středně silných inhibitorů CYP3A4, např. diltiazemu, se doporučuje maximální dávka kombinace ezetimibu a simvastatinu 10/20 mg (viz bod 4.2).
Simvastatin je substrát efluxního transportéru BCRP (Breast Cancer Resistant Protein). Současné podávání přípravků, které jsou inhibitory BCRP (např. elbasvir a grazoprevir), může vést ke zvýšení plazmatické koncentrace simvastatinu a zvýšenému riziku myopatie; proto se má v závislosti na předepsané dávce zvážit úprava dávky simvastatinu. Současné podávání elbasviru a grazopreviru se simvastatinem nebylo studováno, nicméně u pacientů se současnou léčbou přípravky obsahujícími elbasvir nebo grazoprevir nemá dávka ezetimibu/simvastatinu přesáhnout 10/20 mg denně (viz bod 4.5).
Bezpečnost a účinnost kombinace ezetimibu a simvastatinu podávaného současně s fibráty nebyly sledovány. Existuje zvýšené riziko myopatie, jestliže je simvastatin užíván současně s fibráty (zejména s gemfibrozilem). Proto je současné užívání kombinace ezetimibu a simvastatinu s gemfibrozilem kontraindikováno (viz bod 4.3) a současné užívání s jinými fibráty se nedoporučuje (viz bod 4.5).
Jaterní enzymy
V kontrolovaných studiích současného podávání ezetimibu se simvastatinem bylo pozorováno postupné zvyšování koncentrací aminotransferáz. (> 3násobek ULN) (viz bod 4.8).
Ve studii IMPROVE-IT bylo randomizováno 18 144 pacientů s ischemickou chorobou srdeční a akutním koronárním syndromem v anamnéze do skupiny léčené ezetimibem/simvastatinem 10 mg/40 mg denně (n = 9 067) nebo 40 mg simvastatinu denně (n = 9 077). Během mediánu sledování 6,0 roku byla incidence následných zvýšení hladin aminotransferáz (> 3× ULN) 2,5 % u ezetimibu/simvastatinu a 2,3 % u simvastatinu (viz bod 4.8).
-
V klinické studii, ve které bylo randomizováno více než 9 000 pacientů s chronickým onemocněním ledvin do skupiny léčené kombinací ezetimibu a simvastatinu 10/20 mg denně (n = 4 650) nebo placebem (n = 4 620) (medián doby pozorování 4,9 roku), byla incidence následných zvýšení aminotransferáz (> 3 X ULN) 0,7 % u kombinace ezetimibu a simvastatinu a 0,6 % u placeba (viz bod 4.8).
Doporučuje se provést vyšetření jaterní funkce ještě před zahájením léčby kombinací ezetimibu a simvastatinu a dále kdykoli je to z klinického hlediska indikováno. U pacientů titrovaných na 10/80mg dávku je vždy nutno provést další vyšetření před titrací, 3 měsíce po titraci na 10/80mg dávku a dále pravidelně (např. každého půl roku) po dobu prvního roku léčby. Zvláštní pozornost je třeba věnovat pacientům se zvýšenými sérovými koncentracemi aminotransferáz a u těchto pacientů je nutno měření brzy opakovat a pak provádět častěji. Při zjištění progrese zvyšování koncentrací aminotransferáz, zvláště pokud se zvýší na trojnásobek ULN a zvýšení přetrvává, je nutno přípravek vysadit. Mějte na paměti, že ALT se může uvolňovat ze svalů, proto vzestup ALT s CK může ukazovat na myopatii (viz výše myopatie/rhabdomyolýza).
U pacientů užívajících statiny, včetně simvastatinu, existují vzácná poregistrační hlášení fatálního a nefatálního selhání jater. Pokud se během léčby kombinací ezetimibu a simvastatinu objeví závažné poškození jater s klinickými symptomy a/nebo hyperbilirubinemií nebo žloutenkou, léčbu je nutno ihned přerušit. Pokud se nezjistí žádná jiná etiologie, ezetimib/simvastatin znovu nenasazujte.
Ezetimib/simvastatin je nutno podávat opatrně pacientům konzumujícím alkohol ve větším množství.
Porucha funkce jater
Vzhledem k neznámým účinkům zvýšené expozice ezetimibu u pacientů se středně těžkou nebo těžkou poruchou funkce jater se nedoporučuje těmto jedincům ezetimib/simvastatin podávat (viz bod 5.2).
Diabetes mellitus
Některé důkazy naznačují, že statiny zvyšují hladinu glukózy v krvi a u některých pacientů s rizikem vzniku diabetu, mohou vyvolat hyperglykemii, která již vyžaduje diabetologickou péči. Toto riziko však nepřevažuje nad prospěchem léčby statiny – redukcí kardiovaskulárního rizika a není proto důvod pro ukončení léčby statiny. Pacienti se zvýšeným rizikem pro vznik diabetu (glukóza nalačno 5,6 až 6,9 mmol/l, BMI>30kg/m2, zvýšení triacylglycerolů v krvi, hypertenze) mají být klinicky a biochemicky monitorováni v souladu s národními doporučeními.
Pediatrická populace (10 až 17 let věku)
Účinnost a bezpečnost ezetimibu podávaného současně se simvastatinem pacientům ve věku 10 až 17 let s heterozygotní familiární hypercholesterolemií byla hodnocena v kontrolované klinické studii u dospívajících chlapců (Tannerův stupeň II nebo vyšší) a dívek, které již alespoň rok menstruovaly.
-
V této omezené kontrolované studii obecně nebyl u dospívajících chlapců nebo dívek prokázán žádný detekovatelný účinek na růst a pohlavní zrání, ani žádný účinek na menstruační cyklus u dívek. Nicméně účinky ezetimibu na růst a pohlavní zrání při trvání léčby > 33 týdnů nebyly hodnoceny (viz body 4.2 a 4.8).
Bezpečnost a účinnost ezetimibu podávaného současně s dávkami simvastatinu vyššími než 40 mg denně nebyly u pediatrických pacientů ve věku 10 až 17 let hodnoceny.
Ezetimib nebyl u pacientů mladších 10 let nebo dívek před první menstruací hodnocen. (Viz body 4.2 a 4.8.)
Dlouhodobá účinnost léčby ezetimibem u pacientů mladších 17 let s ohledem na snížení morbidity a mortality v dospělosti nebyla hodnocena.
Fibráty
Bezpečnost a účinnost ezetimibu podávaného společně s fibráty nebyly zkoumány (viz výše a body 4.3 a 4.5).
Antikoagulancia
Pokud se přidá ezetimib/simvastatin k warfarinu, jiným kumarinovým antikoagulanciím nebo fluindionu, je nutno řádně sledovat hodnotu International Normalised Ratio (INR) (viz bod 4.5).
Intersticiální plicní choroba
U některých statinů, včetně simvastatinu, byly hlášeny případy intersticiální plicní choroby, zvláště při dlouhodobé léčbě (viz bod 4.8). Příznaky mohou zahrnovat dušnost, neproduktivní kašel a celkové zhoršení zdravotního stavu (únava, úbytek tělesné hmotnosti a horečka). V případě podezření, že se u pacienta vyvinula intersticiální plicní choroba, musí být léčba ezetimibem/simvastatinem vysazena.
Pomocná látka
Tento léčivý přípravek obsahuje monohydrát laktosy. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, úplným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy nemají tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Farmakodynamické interakce
Interakce s hypolipidemiky, které mohou při samostatném podávání vyvolat myopatii
Riziko myopatie včetně rhabdomyolýzy se zvyšuje při současném podávání simvastatinu s fibráty. Navíc dochází k farmakokinetické interakci simvastatinu s gemfibrozilem, která vede ke zvýšení plazmatických koncentrací simvastatinu (viz dále Farmakokinetické interakce a body 4.3 a 4.4). Vzácné případy myopatie/rhabdomyolýzy se vyskytly při současném podávání simvastatinu s niacinem v lipidy modifikujících dávkách (> 1 g/den) (viz bod 4.4).
Fibráty mohou zvýšit vylučování cholesterolu do žluči s následným rozvojem cholelitiázy. V preklinické studii se psy zvýšil ezetimib cholesterol ve žlučníkové žluči (viz bod 5.3). I když význam tohoto preklinického zjištění pro člověka není znám, současné podávání kombinace ezetimibu a simvastatinu a fibrátů se nedoporučuje (viz bod 4.4).
Farmakokinetické interakce
Doporučení pro předepisování pro vzájemně reagující látky jsou shrnuty v tabulce níže (další podrobnosti jsou uvedeny v textu; viz také body 4.2, 4.3 a 4.4).
Lékové interakce související se zvýšeným rizikem myopatie/rhabdomyolýzy
| Reagující látky | Předepsaná doporučení |
| Silné inhibitory CYP3A4, např: Itrakonazol Ketokonazol Posakonazol Vorikonazol Erythromycin Klarithromycin Telithromycin Inhibitory HIV-proteázy (např. nelfinavir) Boceprevir Telaprevir Nefazodon Kobicistat Cyklosporin Danazol Gemfibrozil | Kontraindikováno s kombinací ezetimibu a simvastatinu |
| Jiné fibráty Kyselina fusidová | Nedoporučuje se podávat s kombinací ezetimibu a simvastatinu |
| Niacin (kyselina nikotinová) (> 1 g/den) | Nedoporučuje se podávat s kombinací ezetimibu a simvastatinu u asijských pacientů. |
| Amiodaron Amlodipin Verapamil Diltiazem Niacin (> 1 g/den) Elbasvir Grazoprevir | Nesmí být překročena dávka 10/20 mg kombinace ezetimibu a simvastatinu denně. |
| Lomitapid | U pacientů s HoFH nesmí být překročena dávka 10/40 mg kombinace ezetimibu a simvastatinu denně. |
| Grapefruitová šťáva | Je třeba se vyvarovat požití grapefruitové šťávy během podávání kombinace ezetimibu a simvastatinu |
Působení jiných léčivých přípravků na ezetimib/simvastatin
Kombinace ezetimibu a simvastatinu
Niacin: ve studii na 15 zdravých dospělých způsobilo souběžné podávání kombinace ezetimibu a simvastatinu (10/20 mg denně po dobu 7 dní) mírný vzestup středních hodnot plochy pod křivkou AUC niacinu (22 %) a nikotinylglycinu (19 %) podávaných v přípravku NIASPAN tablety s prodlouženým uvolňováním (1 000 mg po dobu 2 dní a 2 000 mg po dobu 5 dní po nízkotučné snídani). Ve stejné studii současně podávaný přípravek NIASPAN lehce zvyšoval střední hodnoty plochy pod křivkou AUC ezetimibu (9 %), celkového ezetimibu (26 %), simvastatinu (20 %) a kyseliny simvastatinové (35 %) (viz bod 4.2. a 4.4.).
Studie lékových interakcí s vyššími dávkami simvastatinu nebyly provedeny.
Ezetimib
Antacida: Současné podávání antacid snížilo rychlost absorpce ezetimibu, nemělo však žádný vliv na biologickou dostupnost ezetimibu. Tato snížená rychlost absorpce se nepovažuje za klinicky významnou.
Kolestyramin: Současné podávání kolestyraminu zmenšilo průměrnou velikost plochy pod křivkou (area under the curve, AUC) celkového ezetimibu (ezetimib + ezetimib glukuronid) přibližně o 55 %. Postupné snížení LDL-C při přidávání ezetimibu/simvastatinu k kolestyraminu se může touto interakcí zpomalit (viz bod 4.2).
Cyklosporin: Ve studii s osmi pacienty po transplantaci ledvin s clearance kreatininu >50 ml/min, kteří byli na stabilní dávce cyklosporinu, vedlo podávání jednotlivé 10 mg dávky ezetimibu ke 3,4násobnému (rozmezí 2,3–7,9násobné) zvýšení průměrné AUC celkového ezetimibu ve srovnání se zdravou populací z kontrolní skupiny, která dostávala ezetimib samostatně, z jiné studie (n = 17). V další studii se u pacienta po transplantaci ledvin s těžkou poruchou funkce ledvin, který dostával cyklosporin a další mnohonásobnou terapii, projevila 12násobně vyšší expozice celkovému ezetimibu ve srovnání se souběžnými kontrolními skupinami, které dostávaly ezetimib samostatně. V crossover studii ve dvou obdobích, která se provedla se 12 zdravými jedinci, vedlo denní podávání ezetimibu v dávce 20 mg po dobu 8 dní spolu s jednorázovým podáním cyklosporinu v dávce 100 mg sedmý den k průměrnému 15% zvětšení AUC cyklosporinu (rozmezí 10% pokles až 51% zvýšení) ve srovnání s jednorázovým podáním 100mg dávky samotného cyklosporinu. Kontrolovaná studie vlivu současného podávání ezetimibu na expozici cyklosporinu u pacientů po transplantaci ledvin nebyla dosud provedena. Současné podávání kombinace ezetimibu a simvastatinu a cyklosporinu je kontraindikováno (viz bod 4.3).
Fibráty: Současné podávání fenofibrátu nebo gemfibrozilu zvýšilo koncentrace celkového ezetimibu přibližně 1,5násobně a přibližně 1,7násobně v tomto pořadí. I když se uvedená zvýšení nepovažují za klinicky významná, současné podávání kombinace ezetimibu a simvastatinu s gemfibrozilem je kontraindikováno a s jinými fibráty se nedoporučuje (viz body 4.3 a 4.4).
Simvastatin
Simvastatin je substrátem cytochromu P450 3A4. Účinné inhibitory cytochromu P450 3A4 zvyšují riziko myopatie a rhabdomyolýzy tím, že zvyšují plazmatickou koncentraci inhibitorů HMG-CoA reduktázy během léčby simvastatinem. Mezi tyto inhibitory patří itrakonazol, ketokonazol, posakonazol, vorikonazol, erythromycin, klarithromycin, telithromycin, inhibitory HIV-proteázy (např. nelfinavir), boceprevir, telaprevir, nefazodon a léčivé přípravky obsahující kobicistat. Současné podávání itrakonazolu vedlo k více než 10násobnému zvýšení expozice kyselině simvastatinové (aktivní metabolit beta-hydroxykyseliny). Telithromycin vedl k 11násobnému zvýšení expozice kyselině simvastatinové.
Kombinace s itrakonazolem, ketokonazolem, posakonazolem, vorikonazolem, inhibitory HIV- proteázy (např. nelfinavirem), boceprevirem, telaprevirem, erythromycinem, klarithromycinem, telithromycinem, nefazodonem a léčivými přípravky obsahujícími kobicistat je kontraindikována, stejně tak s gemfibrozilem, cyklosporinem a danazolem (viz bod 4.3). Pokud je léčba silnými inhibitory CYP3A4 (látky, které zvyšují AUC přibližně pěti- nebo vícenásobně) nevyhnutelná, je nutno během této léčby podávání kombinace ezetimibu a simvastatinu dočasně přerušit (a zvážit podávání jiného statinu). Opatrnosti je třeba při podávání kombinace ezetimibu a simvastatinu s některými jinými, méně účinnými inhibitory CYP3A4: flukonazolem, verapamilem, diltiazemem (viz body 4.2 a 4.4).
Flukonazol: V souvislosti se současným podáváním simvastatinu a flukonazolu byly hlášeny vzácné případy rhabdomyolýzy. (viz bod 4.4).
Cyklosporin: Riziko rozvoje myopatie/rhabdomyolýzy se při současném podávání cyklosporinu a ezetimibu/simvastatinu zvyšuje. Proto je podávání spolu s cyklosporinem kontraindikováno (viz body 4.3 a 4.4). Přestože není mechanismus zcela objasněn, zdá se, že cyklosporin zvětšuje AUC inhibitorů HMG-CoA reduktázy. Zvětšení velikosti AUC kyseliny simvastatinové, je nejspíše zčásti v důsledku inhibice CYP3A4 a/nebo OATP1B1.
Danazol: Riziko rozvoje myopatie a rhabdomyolýzy se zvyšuje při současném podávání danazolu s ezetimibem/simvastatinem , proto je podávání danazolu kontraindikováno (viz body 4.3 a 4.4).
Gemfibrozil: Gemfibrozil zvětšuje velikost AUC simvastatinové kyseliny 1,9násobně, nejspíše v důsledku blokády dráhy glukuronidace a/nebo OATP1B1 (viz body 4.3 a 4.4). Současné podávání s gemfibrozilem je kontraindikováno.
Kyselina fusidová: Riziko myopatie včetně rhabdomyolýzy se při současném systémovém podávání kyseliny fusidové se statiny může zvyšovat. Mechanismus této interakce (zda jde o interakci farmakodynamickou nebo farmakokinetickou nebo obě) není dosud znám. U pacientů léčených touto kombinací byla hlášena rhabdomyolýza (včetně několika smrtelných případů). Současné podávání této kombinace může vést ke zvýšeným plazmatickým koncentracím obou látek. Pokud je léčba kyselinou fusidovou nezbytná, musí se po dobu léčby kyselinou fusidovou přerušit léčba ezetimibem/simvastatinem (viz bod 4.4.).
Amiodaron: Riziko myopatie a rhabdomyolýzy se zvyšuje při současném podávání amiodaronu se simvastatinem (viz bod 4.4). V klinické studii byla myopatie hlášena u 6 % pacientů léčených simvastatinem v dávce 80 mg a amiodaronem. Proto nesmí dávka ezetimibu/simvastatinu u pacientů současně léčených amiodaronem přesáhnout 10/20 mg denně.
Blokátory vápníkového kanálu
Verapamil
Riziko myopatie a rhabdomyolýzy je současným podáváním verapamilu se simvastatinem v dávce 40 nebo 80 mg zvýšeno (viz bod 4.4). Ve farmakokinetické studii vedlo současné podávání simvastatinu a verapamilu k 2,3násobnému zvýšení expozice kyselině simvastatinové, zčásti nejspíše v důsledku inhibice CYP3A4. Dávka ezetimibu/simvastatinu proto nesmí překročit hodnotu 10/20 mg denně u pacientů užívajících současně verapamil.
Diltiazem
Riziko myopatie a rhabdomyolýzy je současným podáváním diltiazemu se simvastatinem v dávce 80 mg zvýšeno (viz bod 4.4). Ve farmakokinetické studii vyvolalo současné podávání diltiazemu a simvastatinu 2,7násobné zvýšení expozice kyselině simvastatinové, pravděpodobně v důsledku inhibice CYP3A4. Dávka ezetimibu/simvastatinu proto nesmí překročit hodnotu 10/20 mg denně u pacientů současně užívajících diltiazem.
Amlodipin
Pacienti, kterým se podává amlodipin, léčení současně simvastatinem jsou vystaveni vyššímu riziku myopatie. Ve farmakokinetické studii způsobilo současné podání amlodipinu 1,6násobné zvýšení expozice kyselině simvastatinové. Proto u pacientů současně léčených amlodipinem nesmí dávka ezetimibu/simvastatinu přesáhnout 10/20 mg denně.
Lomitapid
Při současném podávání simvastatinu s lomitapidem může být riziko myopatie a rhabdomyolýzy zvýšeno (viz body 4.3 a 4.4). Proto u pacientů s HoFH, kteří užívají současně lomitapid, nesmí dávka ezetimibu/simvastatinu překročit 10/40 mg denně.
Středně silné inhibitory CYP3A4
Pacienti užívající spolu s kombinací ezetimibu a simvastatinu, zejména kombinace ezetimibu a simvastatinu obsahující vyšší dávky, jiná léčiva, u nichž je uvedeno, že mají středně silný inhibiční účinek na CYP3A4, mohou být vystaveni vyššímu riziku myopatie (viz bod 4.4).
Inhibitory transportního proteinu OATP1B1
Kyselina simvastatinová je substrát transportního proteinu OATP1B1. Současné podávání léčivých přípravků, které jsou inhibitory transportního proteinu OATP1B1, může vést ke zvýšeným plazmatickým koncentracím kyseliny simvastatinové a ke zvýšenému riziku myopatie (viz body 4.3 a 4.4).
Inhibitory BCRP (Breast Cancer Resistant Protein)
Současné podávání léčivých přípravků, které jsou inhibitory BCRP, včetně přípravků obsahujících elbasvir nebo grazoprevir, může vést ke zvýšení plazmatické koncentrace simvastatinu a zvýšenému riziku myopatie (viz body 4.2 a 4.4).
Grapefruitová šťáva
Grapefruitová šťáva inhibuje cytochrom P450 3A4. Současná konzumace velkých množství (nad 1 litr denně) grapefruitové šťávy a simvastatinu vedla k 7násobnému zvýšení expozice kyselině simvastatinové. Příjem 240 ml grapefruitové šťávy ráno a podání simvastatinu večer také vedlo k 1,9násobnému zvýšení. Konzumaci grapefruitové šťávy v průběhu léčby ezetimibem/simvastatinem je proto nutno se vyvarovat.
Kolchicin
Při současném podávání kolchicinu a simvastatinu se objevila hlášení myopatie a rhabdomyolýzy, a to u pacientů s poruchou funkce ledvin. U pacientů užívajících tuto kombinaci se doporučuje pečlivé klinické sledování.
Rifampicin
Jelikož je rifampicin silným induktorem CYP3A4, může se u pacientů dlouhodobě léčených rifampicinem (např. léčba tuberkulózy) objevit ztráta účinku simvastatinu. Ve farmakokinetické studii na normálních dobrovolnících byla při současném podání rifampicinu plocha pod křivkou průběhu koncentrace kyseliny simvastatinové v plazmě (AUC) snížena o 93 %.
Niacin
Byly pozorovány případy myopatie/rhabdomyolýzy při současném podávání simvastatinu a lipidy modifikujících dávek (> 1 g/den) niacinu (viz bod 4.4).
Účinky kombinace ezetimibu a simvastatinu na farmakokinetiku jiných léčivých přípravků
Ezetimib
-
V preklinických studiích se ukázalo, že ezetimib neindukuje enzymy cytochromu P450 metabolizující léčivé látky. Nebyly pozorovány žádné klinicky významné farmakokinetické interakce mezi ezetimibem a léčivými látkami, o nichž je známo, že se metabolizují cytochromy P450 1A2, 2D6, 2C8, 2C9 a 3A4 nebo N-acetyltransferázou.
Antikoagulancia
Ve studii s 12 zdravými dospělými muži nemělo současné podávání ezetimibu (10 mg jednou denně) žádný statisticky významný vliv na biologickou dostupnost warfarinu a protrombinový čas.
-
V postmarketingových hlášeních se však objevily zprávy o zvýšených hodnotách International Normalised Ratio (INR) u pacientů, u nichž byl ezetimib přidán k warfarinu nebo fluindionu. V případech, kdy se ezetimib/simvastatin přidá k warfarinu, jinému kumarinovému antikoagulanciu nebo fluindionu, je nutno INR řádně sledovat (viz bod 4.4).
Simvastatin
Simvastatin nemá na cytochrom P450 3A4 inhibiční účinek. Neočekává se tedy, že by simvastatin ovlivňoval plazmatické koncentrace látek biotransformovaných cytochromem P450 3A4.
Perorální antikoagulancia
Ve dvou klinických studiích, jedné se zdravými dobrovolníky a druhé s pacienty s hypercholesterolemií, simvastatin v dávkách 20–40 mg/denně mírně potencoval účinek kumarinových antikoagulancií: protrombinový čas, uváděný jako International Normalised Ratio (INR), se prodloužil z výchozí hodnoty 1,7 na 1,8 ve studii s dobrovolníky a z 2,6 na 3,4 ve studii s pacienty. Byly popsány velmi vzácné případy zvýšení INR. U pacientů užívajících kumarinová antikoagulancia je třeba před zahájením léčby ezetimibem/simvastatinem stanovit protrombinový čas a poté ho stanovovat dostatečně často v počátcích léčby, aby byla jistota, že nedochází k významným změnám protrombinového času. Po stabilizaci protrombinového času jej lze sledovat v intervalech obvykle doporučovaných pro pacienty užívající kumarinová antikoagulancia. Tento postup je třeba opakovat při změně dávky ezetimibu/simvastatinu nebo jeho vysazení. U pacientů, kteří neužívali antikoagulancia, nebyla léčba simvastatinem spojována s krvácením nebo se změnami protrombinového času.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Ateroskleróza je chronický děj a za normálních okolností by vysazení hypolipidemických léčivých přípravků během těhotenství mělo mít minimální dopad na dlouhodobé riziko související s primární hypercholesterolemií.
Kombinace ezetimibu a simvastatinu
Během těhotenství je kombinace ezetimibu a simvastatinu kontraindikována. O užívání kombinace ezetimibu a simvastatinu během těhotenství nejsou k dispozici žádné klinické údaje. Studie kombinační terapie u zvířat prokázaly reprodukční toxicitu. (Viz bod 5.3.)
Simvastatin
Bezpečnost simvastatinu u těhotných žen nebyla hodnocena. U těhotných žen se neprováděly žádné kontrolované klinické studie se simvastatinem. Vzácně se objevily zprávy o vrozených anomáliích po nitroděložní expozici inhibitorům HMG-CoA reduktázy. V analýze přibližně 200 prospektivně sledovaných těhotenství s expozicí simvastatinu nebo jinému blízce příbuzného inhibitoru HMG-CoA reduktázy v prvním trimestru byla incidence vrozených anomálií srovnatelná s hodnotou pozorovanou u celkové populace. Uvedený počet těhotenství byl ze statistického hlediska dostatečný k vyloučení 2,5násobného nebo většího nárůstu vrozených anomálií ve srovnání s běžnou incidencí.
Přestože nejsou k dispozici důkazy o tom, že by se výskyt vrozených anomálií u potomstva pacientů užívajících simvastatin nebo jiný blízce příbuzný inhibitor HMG-CoA reduktázy lišil od hodnot pozorovaných u celkové populace, může podávání simvastatinu nastávající matce snížit koncentrace mevalonátu u plodu; mevalonát je prekurzorem biologické syntézy cholesterolu. Z tohoto důvodu se nesmí kombinace ezetimibu a simvastatinu podávat těhotným ženám, ženám, které se snaží otěhotnět nebo ženám, které se domnívají, že jsou těhotné. Léčbu kombinací ezetimibu a simvastatinu je třeba vysadit na dobu těhotenství nebo do ověření, že žena není těhotná. (Viz bod 4.3.)
Ezetimib
O užívání ezetimibu během těhotenství nejsou k dispozici žádné klinické údaje.
Kojení
Během kojení je kombinace ezetimibu a simvastatinu kontraindikována. Studie s potkany prokázaly, že ezetimib se vylučuje do mateřského mléka. Není známo, zda se léčivé látky kombinace ezetimibu a simvastatinu vylučují do mateřského mléka. (Viz bod 4.3.)
Fertilita
Ezetimib
Nejsou k dispozici žádné údaje z klinických studií týkající se vlivu ezetimibu na fertilitu člověka. Ezetimib nemá žádný vliv na fertilitu samců nebo samic potkanů (viz bod 5.3).
Simvastatin
Nejsou k dispozici žádné údaje z klinických studií týkající se vlivu simvastatinu na fertilitu člověka. Simvastatin nemá žádný vliv na fertilitu samců nebo samic potkanů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit a používat stroje nebyly provedeny. Při řízení vozidel nebo ovládání strojů je však třeba vzít v úvahu, že byly hlášeny závratě.
4.8
Nežádoucí účinky
Tabulkový seznam nežádoucích účinků (klinické studie)
Ezetimib/simvastatin (nebo současné podávání ezetimibu a simvastatinu odpovídající kombinaci ezetimibu a simvastatinu) byl hodnocen z hlediska bezpečnosti v klinických studiích přibližně u 12 000 pacientů.
Častost výskytu nežádoucích účinků je seřazena následujícím způsobem: Velmi časté (>1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit) včetně izolovaných případů.
Následující nežádoucí účinky byly pozorovány u pacientů léčených kombinací ezetimibu a simvastatinu (n= 2 404) a s vyšší incidencí než u placeba (n= 1 340).
| Nežádoucí účinky kombinace ezetimibu a simvastatinu vyskytující se s vyšší incidencí než u placeba | ||
| Třída orgánových systémů | Časté | Méně časté |
| Psychiatrické poruchy | poruchy spánku | |
| Poruchy nervového systému | závrať; bolest hlavy | |
| Gastrointestinální poruchy | bolest břicha; břišní diskomfort; bolest v horní části břicha; dyspepsie; flatulence; nauzea; zvracení | |
| Poruchy kůže a podkožní tkáně | pruritus, vyrážka | |
| Poruchy svalové a kosterní soustavy a pojivové tkáně | artralgie; svalové spasmy; svalová slabost muskuloskeletální diskomfort; bolest krku; bolest končetin | |
| Celkové poruchy a reakce v místě aplikace | astenie; únava; malátnost; periferní edém | |
| Vyšetření | Zvýšení ALT a/nebo AST Vzestup kreatinkinázy v krvi | zvýšení bilirubinu v krvi, zvýšení kyseliny močové v krvi; zvýšení gama-glutamyltransferázy; zvýšení INR; přítomnost bílkoviny v moči; snížení tělesné hmotnosti |
Následující nežádoucí účinky byly pozorovány u pacientů léčených kombinací ezetimibu a simvastatinu (n = 9 595) a s vyšší incidencí než u statinů podávaných samotných (n = 8 883).
| Nežádoucí účinky kombinace ezetimibu a simvastatinu vyskytující se s vyšší incidencí než u statinů | ||
| Třída orgánových systémů | Časté | Méně časté |
| Psychiatrické poruchy | insomnie | |
| Poruchy nervového systému | bolest hlavy; parestezie | |
| Gastrointestinální poruchy | břišní distenze; průjem; sucho v ústech; dyspepsie; flatulence; refluxní choroba jícnu; zvracení | |
| Poruchy kůže a podkožní tkáně | pruritus; vyrážka; kopřivka | |
| Poruchy svalové a kosterní soustavy a pojivové tkáně | myalgie | artralgie; bolest zad; svalové spasmy; svalová slabost; muskuloskeletální bolest; bolest |
| končetin | ||
| Celkové poruchy a reakce v místě aplikace | astenie; bolest na hrudi; únava; periferní edém | |
| Vyšetření | zvýšení ALT a/nebo AST | zvýšení bilirubinu v krvi; zvýšení kreatinkinázy v krvi; zvýšení gammaglutamyltransferázy |
Pediatrická populace (od 10 do 17 let věku)
Ve studii zahrnující dospívající (10 až 17 let věku) pacienty s heterozygotní familiární hypercholesterolemií (n = 248) bylo u 3 % (4 pacienti) pacientů léčených kombinací ezetimibu a simvastatinu zjištěno zvýšení ALT a/nebo AST (> 3násobek horní hranice normálu, několikrát po sobě) v porovnání se 2 % (2 pacienti) ve skupině léčené simvastatinem v monoterapii; tato čísla byla ohledně zvýšení kreatinfosfokinázy (CPK) 2 % (2 pacienti), respektive 0 % (> 10násobek horní hranice normálu). Nebyly hlášeny žádné případy myopatie.
Hodnocení nebylo vhodné pro porovnání vzácných nežádoucích účinků.
Pacienti s ischemickou chorobou srdeční a akutním koronárním syndromem v anamnéze
Ve studii IMPROVE-IT (viz bod 5.1), která zahrnovala 18 144 pacientů léčených ezetimibem/simvastatinem 10 mg/40 mg (n = 9 067, z toho 6 % bylo titrováno na ezetimib/simvastatin 10 mg/80 mg) nebo simvastatinem v dávce 40 mg (n = 9 077, z toho 27 % bylo titrováno na 80 mg simvastatinu), byly bezpečnostní profily během mediánu sledování 6,0 roku podobné. Kvůli výskytu nežádoucích účinků ukončilo léčbu 10,6 % pacientů léčených ezetimibem/simvastatinem a 10,1 % pacientů léčených simvastatinem. Incidence myopatie byla u pacientů s ezetimibem/simvastatinem 0,2 % a u pacientů se simvastatinem 0,1 %, přičemž myopatie byla definována jako nevysvětlená svalová slabost nebo bolest s hladinou sérové CK > 10násobek ULN nebo s koncentrací CK > 5násobek a zároveň < 10násobek ULN ve dvou následných měřeních. Incidence rhabdomyolýzy byla u pacientů s ezetimibem/simvastatinem 0,1 % a u pacientů se simvastatinem 0,2 %, přičemž rhabdomyolýza byla definována jako nevysvětlená svalová slabost nebo bolest s hladinou sérové CK > 10násobek ULN s prokázaným renálním poškozením, > 5násobek a zároveň < 10násobek ULN ve dvou následných měřeních s prokázaným renálním poškozením nebo > 10 000 IU/l bez prokázaného renálního poškození. Incidence následných zvýšení hladin aminotransferáz (> 3× ULN) byla 2,5 % u ezetimibu/simvastatinu a 2,3 % u simvastatinu (viz bod 4.4.). Nežádoucí účinky související se žlučníkem byly hlášeny u 3,1 % pacientů léčených ezetimibem/simvastatinem a u 3,5 % pacientů léčených simvastatinem. Incidence hospitalizace kvůli cholecystektomii byla v obou skupinách 1,5 %. Rakovina (definovaná jako jakákoli nová malignita) byla během studie diagnostikována u 9,4 % pacientů léčených
ezetimibem/simvastatinem oproti 9,5 % pacientů léčených simvastatinem.
Pacienti s chronickým onemocněním ledvin
Ve studii „Study of Heart and Renal Protection“ (SHARP) (viz bod 5.1), která zahrnovala více než 9 000 pacientů léčených kombinací ezetimibu a simvastatinu 10/20 mg denně (n = 4 650) nebo placebem (n = 4 620), byly bezpečnostní profily srovnatelné v průběhu mediánu doby sledování 4,9 roku. V této studii byly zaznamenávány pouze závažné nežádoucí účinky a ukončení léčby kvůli případným nežádoucím účinkům. Míry ukončení léčby v důsledku nežádoucích účinků byly srovnatelné (10,4 % u pacientů léčených ezetimibem/simvastatinem, 9,8 % u pacientů léčených placebem). Incidence myopatie/rhabdomyolýzy byla 0,2 % u pacientů léčených ezetimibem/simvastatinem a 0,1 % u pacientů léčených placebem. Následné zvýšení aminotransferáz (> 3 X ULN) se vyskytlo u 0,7 % pacientů léčených ezetimibem/simvastatinem v porovnání s 0,6 % pacientů léčených placebem. V této studii nebyla žádná statisticky významná zvýšení incidence předem specifikovaných nežádoucích účinků, včetně rakoviny (9,4 % u ezetimibu/simvastatinu, 9,5 % u placeba), hepatitidy, cholycystektomie nebo komplikací žlučníkových kamenů či pankreatitidy.
Laboratorní hodnoty
Ve studiích současného podávání kombinace ezetimibu a simvastatinu s jinými přípravky dosáhla incidence klinicky významných zvýšení koncentrací sérových aminotransferáz (ALT a/nebo AST > 3násobek ULN, následující po sobě) u pacientů léčených ezetimibem/simvastatinem 1,7 %. Tato zvýšení byla obecně asymptomatická, nebyla spojena s rozvojem cholestázy a po vysazení terapie nebo při pokračování léčby se koncentrace vrátily na výchozí hodnoty (viz bod 4.4).
Klinicky významná zvýšení hodnot CK (> IQxULN) byla pozorována u 0,2 % pacientů užívajících ezetimib/simvastatin.
Zkušenosti po uvedení na trh
Následující nežádoucí účinky byly hlášeny během postmarketingového použití kombinace ezetimibu a simvastatinu nebo během klinických studií nebo během postmarketingovém použití každé z jednotlivých složek kombinace ezetimibu a simvastatinu.
| Není známo | |
| Poruchy krve a lymfatického systému |
|
| Poruchy metabolismu a výživy |
|
| Psychiatrické poruchy |
|
| Poruchy nervového systému |
|
| Cévní poruchy |
|
| Respirační, hrudní a mediastinální poruchy |
|
| Gastrointestinální poruchy |
|
| Poruchy jater a žlučových cest |
|
| Poruchy kůže a podkožní tkáně |
|
| Poruchy imunitního systému |
|
| Poruchy svalové a kosterní soustavy a pojivové tkáně |
|
| Poruchy reprodukčního systému a prsu |
|
| Celkové poruchy a reakce v místě aplikace |
|
* V klinické studii se myopatie vyskytovala často u pacientů léčených simvastatinem v dávce 80 mg/den v porovnání s pacienty léčenými dávkou 20 mg/den (1,0 % vs 0,02 %, v uvedeném pořadí) (viz body 4.4 a 4.5).
Velmi vzácně byla hlášena imunitně zprostředkovaná nekrotizující myopatie (IMNM), autoimunitní myopatie spojovaná s užíváním statinů. Pro IMNM je charakteristické: slabost proximálních svalových skupin a zvýšení kreatinkinázy v séru, které přetrvává navzdory vysazení léčby statiny; biopsie svalů ukazující nekrotizující myopatii bez významného zánětu; zlepšení po použití imunosupresivních látek (viz bod 4.4).
Zřejmý syndrom hypersenzitivity byl popsán vzácně, byly při něm pozorovány následující stavy: angioedém, lupusu podobný syndrom, revmatická polymyalgie, dermatomyozitida, vaskulitida, trombocytopenie, eozinofilie, zvýšená sedimentace erytrocytů, artritida a artralgie, kopřivka, fotosenzitivní reakce, pyrexie, zrudnutí, dyspnoe a malátnost.
Laboratorní hodnoty: zvýšení hodnoty alkalické fosfatázy, abnormální hodnoty jaterních testů.
U statinů, včetně simvastatinu, byla hlášena zvýšení hladin HbA1c a sérové glukózy nalačno.
V souvislosti s používáním statinů, včetně simvastatinu, byly zaznamenány vzácné poregistrační hlášení týkající se zhoršení kognitivních schopností (např. ztráta paměti, zapomnětlivost, amnézie, zhoršení paměti, zmatenost). Tato hlášení jsou obecně nezávažná a reverzibilní při vysazení statinů s proměnlivou dobou do nástupu (1 den až roky) a vymizení (medián 3 týdny) symptomů.
U některých statinů byly hlášeny následující nežádoucí účinky:
- Poruchy spánku, včetně nočních můr
- Sexuální dysfunkce
- Diabetes mellitus: Frekvence výskytu bude záviset na přítomnosti nebo absenci rizikových faktorů (glukóza nalačno > 5,6 mmol/l, BMI > 30 kg/m2, zvýšení triacylglycerolů v krvi, hypertenze v anamnéze).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky: http:/
4.9 Předávkování
Ezetimib/simvastatin
V případě předávkování je nutno přijmout symptomatická a podpůrná opatření. V akutních studiích perorální toxicity na myších a potkanech bylo současné podávání ezetimibu (1 000 mg/kg) a simvastatinu (1 000 mg/kg) dobře snášeno. U těchto zvířat nebyly pozorovány klinické známky toxicity. Odhadnutá hodnota perorální LD50 pro oba druhy pro ezetimib činila > 1 000 mg/kg a pro simvastatin > 1 000 mg/kg.
Ezetimib
Simvastatin
Bylo popsáno několik případů předávkování; maximální požitá dávka byla 3,6 g. Všichni pacienti se zotavili bez následků.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: inhibitory HMG-CoA reduktázy v kombinaci s jinými lipidy upravujícími látkami,
ATC kód: C10BA02
Ezetimib/simvastatin je hypolipidemikum, které selektivně blokuje střevní vstřebávání cholesterolu a příbuzných rostlinných sterolů a inhibuje endogenní syntézu cholesterolu.
Mechanismus účinku
Ezetimib/simvastatin
Plazmatický cholesterol vzniká intestinální absorpcí a endogenní syntézou. Kombinace ezetimibu a simvastatinu obsahuje ezetimib a simvastatin, dvě hypolipidemické látky se vzájemně se doplňujícími mechanismy účinku. Ezetimib/simvastatin snižuje zvýšený celkový cholesterol (total-C), LDL-C, apolipoprotein B (Apo B), triacylglyceroly (TG) a cholesterol nevysokodenzitních lipoproteinů (non-HDL-C) a zvyšuje cholesterol vysokodenzitních lipoproteinů (HDL-C) dvojitou inhibicí absorpce a syntézy cholesterolu.
Ezetimib
Ezetimib inhibuje intestinální absorpci cholesterolu. Ezetimib je účinný po perorálním podání a má mechanizmus účinku, který se liší od jiných skupin látek snižujících cholesterol (např. statinů, sekvestrantů žlučových kyselin (pryskyřic), derivátů kyseliny fibrové a rostlinných sterolů). Molekulárním cílem ezetimibu je sterolový přenašeč Niemann-Pick C1 Like 1 (NPC1L1), který je odpovědný za intestinální příjem cholesterolu a fytosterolů.
Ezetimib je lokalizován na kartáčovém lemu tenkého střeva a blokuje absorpci cholesterolu; výsledkem je snížený odvod intestinálního cholesterolu do jater; statiny snižují syntézu cholesterolu v játrech a společně tyto rozdílné mechanismy zajišťují komplementární snižování cholesterolu. Ve 2týdenní klinické studii s 18 pacienty s hypercholesterolemií vykazoval ezetimib sníženou intestinální absorpci cholesterolu o 54 % ve srovnání s placebem.
Byla provedena řada preklinických studií s cílem stanovit selektivitu ezetimibu při blokádě absorpce cholesterolu. Ezetimib inhiboval vstřebávání [14C]-cholesterolu bez vlivu na absorpci triacylglycerolů, mastných kyselin, žlučových kyselin, progesteronu, ethinylestradiolu nebo v tucích rozpustných vitamínů A a D.
Simvastatin
Po perorálním podání je simvastatin, což je inaktivní lakton, hydrolyzován v játrech na příslušnou formu aktivní P-hydroxykyseliny, která účinně inhibuje HMG-CoA reduktázu (3-hydroxy-3-methylglutarylkoenzym A reduktázu). Tento enzym katalyzuje konverzi HMG-CoA na mevalonát, což je časný a rychlost určující krok v biosyntéze cholesterolu.
Ukázalo se, že simvastatin snižuje normální i zvýšené hladiny LDL-C. LDL se tvoří z lipoproteinu o velmi nízké hustotě (VLDL) a k jeho katabolizmu dochází převážně prostřednictvím LDL receptorů s vysokou afinitou. Mechanizmus snižování LDL simvastatinem může zahrnovat jak snižování hladin VLDL-cholesterolu (VLDL-C), tak i indukci receptoru LDL s výsledným snížením produkce a zvýšeným katabolizmem LDL-C. Během léčby simvastatinem dochází i k podstatnému snížení hladin apolipoproteinu B. Kromě toho simvastatin mírně zvyšuje hladiny HDL-C a snižuje plazmatické hladiny TG. V důsledku těchto změn dochází ke snížení poměrů total-C/HDL-C a LDL-C/HDL-C.
Klinická účinnost a bezpečnost
V kontrolovaných klinických studiích snižoval ezetimib/simvastatin statisticky významně celkový cholesterol, LDL-C, Apo B, TG a non-HDL-C a zvyšoval HDL-C u pacientů s hypercholesterolemií.
Prevence kardiovaskulárních příhod
IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) byla multicentrická randomizovaná dvojitě zaslepená studie s aktivním komparátorem, do níž bylo zařazeno 18 144 pacientů během 10 dnů po hospitalizaci kvůli akutnímu koronárnímu syndromu (AKS; buď infarktu myokardu, nebo nestabilní angině pectoris). Hladina LDL-C při projevení AKS byla < 125 mg/dl (< 3,2 mmol/l) u pacientů, kteří neužívali hypolipidemickou léčbu, nebo < 100 mg/dl (< 2,6 mmol/l) u pacientů, kteří dostávali hypolipidemickou léčbu. Všichni pacienti byli v poměru 1:1 randomizováni do skupiny léčené ezetimibem/simvastatinem 10/40 mg (n = 9 067) nebo simvastatinem 40 mg (n = 9 077) a sledováni po medián doby sledování 6,0 roku.
Průměrný věk pacientů byl 63,6 roku, 76 % tvořili muži, 84 % byli běloši a 27 % pacientů mělo diabetes mellitus. Průměrná hodnota LDL-C při příhodě, která byla kvalifikující pro zařazení do studie, byla 80 mg/dl (2,1 mmol/l) u pacientů, kteří dostávali hypolipidemickou léčbu (n = 6 390), a 101 mg/dl (2,6 mmol/l) u těch, kteří předtím nedostávali hypolipidemickou léčbu (n = 11 594). Před hospitalizací kvůli příhodě AKS kvalifikující ke vstupu do studie užívalo 34 % pacientů statin. Po jednom roce byla průměrná hodnota LDL-C u pacientů, kteří pokračovali v léčbě, 53,2 mg/dl (1,4 mmol/l) ve skupině s ezetimibem/simvastatinem a 69,9 mg/dl (1,8 mmol/l) ve skupině se samotným simvastatinem. Hladiny lipidů byly obecně získány u pacientů, kteří pokračovali v léčbě v rámci studie.
Primárním cílovým ukazatelem byl složený ukazatel zahrnující úmrtí z kardiovaskulárních příčin, velké koronární příhody (definované jako nefatální infarkt myokardu, popsaná nestabilní angina pectoris vyžadující hospitalizaci, nebo jakákoli koronární revaskularizace prováděná nejméně 30 dní po randomizaci) a nefatální cévní mozkovou příhodu. Studie prokázala, že léčba ezetimibem spolu se simvastatinem ve srovnání se samotným simvastatinem poskytuje rostoucí přínos ve formě snížení výskytu primárního cílového ukazatele složeného z úmrtí z kardiovaskulárních příčin, velké koronární příhody a nefatální cévní mozkové příhody (relativní snížení rizika je 6,4 %, p = 0,016). Primární cílový ukazatel se vyskytl u 2 572 z 9 067 pacientů ze skupiny ezetimib/simvastatin (pravděpodobnost výskytu za 7 let byla dle Kaplan-Meierovy (KM) metody 32,72 %) a u 2 742 z 9 077 pacientů ze skupiny se samotným simvastatinem (pravděpodobnost výskytu za 7 let byla dle KM metody 34,67 %). (Viz Graf 1 a Tabulka 1.) Očekává se, že tento rostoucí přínos bude podobný při společném podávání s jinými statiny vykazujícími účinek na snižování rizika kardiovaskulárních příhod. Celková úmrtnost se v této vysoce rizikové skupině nezměnila (viz Tabulka 1).
Ve studii byl pozorován celkový přínos pro všechny typy cévní mozkové příhody, nicméně bylo zaznamenáno malé nesignifikantní zvýšení rizika u hemoragické cévní mozkové příhody ve skupině s ezetimibem a simvastatinem ve srovnání se skupinou se samotným simvastatinem (viz Tabulka 1). Riziko hemoragické cévní mozkové příhody pro ezetimib podávaný se statiny s vyšší účinností nebylo hodnoceno v dlouhodobých studiích.
Léčebný účinek kombinace ezetimib/simvastatin byl obecně konzistentní napříč celkovými výsledky mnoha podskupin, dělených podle pohlaví, věku, rasy, diabetu mellitu v anamnéze, počáteční hladiny lipidů, předchozí léčby statiny, cévní mozkové příhody v anamnéze a hypertenze.
Graf 1: Účinek ezetimibu/simvastatinu na primární cílový ukazatel složený z úmrtí z kardiovaskulárních příčin, velké koronární příhody nebo nefatální cévní mozkové příhody
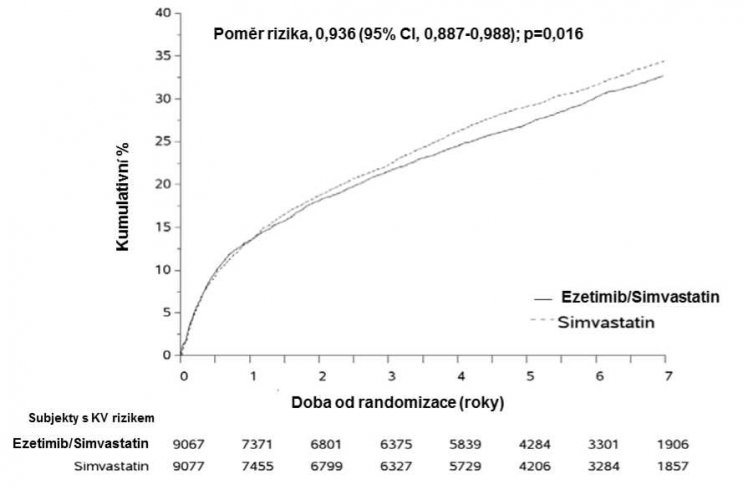
Tabulka 1: Velké kardiovaskulární příhody podle skupin u všech pacientů randomizovaných v IMPROVE-IT
| Výsledek | Ezetimib/Simvastatin 10/40 mga (N=9067) | Simvastatin 40 mgb (N=9077) | Poměr rizika (95% CI) | p-hodnota | ||
| n | K-M%c | n | K-M%c | |||
| Primární složený cílový ukazatel účinnosti | ||||||
| (Úmrtí z kardiovaskulárních příčin, velké koronární příhody a nefatální cévní mozková příhoda) | 2572 | 32,72 % | 2742 | 34,67 % | 0,936 (0,887; 0,988) | 0,016 |
| Sekundární složený cílový ukazatel účinnosti | ||||||
| Úmrtí z důvodu ICHS, nefatální infarkt myokardu, urgentní koronární revaskularizace po 30 dnech | 1322 | 17,52 % | 1448 | 18,88 % | 0,912 (0,847; 0,983) | 0,016 |
| Velká koronární příhoda, nefatální cévní mozková příhoda, úmrtí (z jakékoli příčiny) | 3089 | 38,65 % | 3246 | 40,25 % | 0,948 (0,903; 0,996) | 0,035 |
| Úmrtí z kardiovaskulárních příčin, nefatální infarkt myokardu, nestabilní angina pectoris vyžadující hospitalizaci, jakákoli revaskularizace, nefatální cévní mozková příhoda | 2716 | 34,49 % | 2869 | 36,20 % | 0,945 (0,897; 0,996) | 0,035 |
| Složky primárního složeného cílového ukazatele a vybrané cílové ukazatele účinnosti (první výskyt dané příhody v jakékoli chvíli) | ||||||
| Úmrtí z kardiovaskulárních příčin | 537 | 6,89 % | 538 | 6,84 % | 1,000 (0,887; 1,127) | 0,997 |
| Velká koronární příhoda: | ||||||
| nefatální infarkt myokardu | 945 | 12,77 % | 1083 | 14,41 % | 0,871 (0,798, 0,950) | 0,002 |
| nestabilní angina pectoris vyžadující hospitalizaci | 156 | 2,06 % | 148 | 1,92 % | 1,059 (0,846; 1,326) | 0,618 |
| koronární revaskularizace po 30 dnech | 1690 | 21,84 % | 1793 | 23,36 % | 0,947 (0,886; 1,012) | 0,107 |
| Nefatální cévní mozková příhoda | 245 | 3,49 % | 305 | 4,24 % | 0,802 (0,678, 0,949) | 0,010 |
| Infarkt myokardu (fatální i nefatální) | 977 | 13,13 % | 1118 | 14,82 % | 0,872 (0,800, 0,950) | 0,002 |
| Cévní mozková příhoda (fatální i nefatální) | 296 | 4,16 % | 345 | 4,77 % | 0,857 (0,734, 1,001) | 0,052 |
| nehemoragická cévní mozková příhodad | 242 | 3,48 % | 305 | 4,23 % | 0,793 (0,670, 0,939) | 0,007 |
| hemoragická cévní mozková příhoda | 59 | 0,77 % | 43 | 0,59 % | 1,377 (0,930, 2,040) | 0,110 |
| Úmrtí z jakékoli příčiny | 1215 | 15,36 % | 1231 | 15,28 % | 0,989 (0,914, 1,070) | 0,782 |
a 6 % pacientů bylo titrováno na ezetimib/simvastatin 10/80 mg
b 27 % pacientů bylo titrováno na simvastatin 80 mg
c Kaplan-Meierův odhad pro 7 let
d zahrnuje ischemickou cévní mozkovou příhodu nebo cévní mozkovou příhodu neurčeného typu
Primární hypercholesterolemie
V dvojitě zaslepené, placebem kontrolované, 8týdenní studii bylo 240 pacientů s hypercholesterolemií, kteří již dostávali simvastatin v monoterapii a nedosáhli cílové hodnoty LDL-C podle Národního programu pro osvětu ve snižování cholesterolu (National Cholesterol Education Program, NCEP) (2,6 až 4,1 mmol/l [100 až 160 mg/dl], podle výchozích charakteristik) randomizováno do skupin, které dostávaly buď ezetimib 10 mg nebo placebo navíc k již probíhající léčbě simvastatinem. Z pacientů léčených simvastatinem, kteří při výchozím vyšetření nedosáhli cílové hodnoty LDL-C
(přibližně 80 %), dosáhlo statisticky významně více (76 %) pacientů randomizovaných do skupiny se současným užíváním ezetimibu a simvastatinu cílové hodnoty LDL-C při hodnoceném parametru studie ve srovnání s nemocnými randomizovanými do skupiny se současným užíváním placeba se simvastatinem (21,5 %).
Odpovídající snížení LDL-C při užívání ezetimibu nebo placeba současně podávaných se simvastatinem se také statisticky významně lišilo (27 % nebo 3 %, v uvedeném pořadí). Navíc ezetimib podávaný současně se simvastatinem významně snížil total-C, Apo B a TG ve srovnání s placebem podávaným spolu se simvastatinem.
V multicentrické, dvojitě zaslepené, 24týdenní studii bylo 214 pacientů s diabetem mellitem 2. typu, léčených thiazolindiony (rosiglitazon nebo pioglitazon) po dobu minimálně 3 měsíců a simvastatinem 20 mg po minimální dobu 6 týdnů s průměrnou koncentrací LDL-C 2,4 mmol/l (93 mg/dl), randomizováno do skupin užívajících buď simvastatin 40 mg nebo současně podávané účinné látky v poměru použitém v kombinaci ezetimibu a simvastatinu 10 mg/20 mg. Ezetimib/simvastatin 10 mg/20 mg byl statisticky významně účinnější než zdvojnásobení dávky simvastatinu na 40 mg při dalším snižování LDL-C (-21 % a 0 %), total-C (-14 % a –1 %), Apo B (-14 % a –2 %) a non-HDL-C (-20 % a –2 %, vždy v uvedeném pořadí) pod hodnotu pozorovanou při užívání simvastatinu v dávce 20 mg. Výsledky pro HDL-C a TG u obou léčebných skupin se významně nelišily. Výsledky nebyly ovlivněny typem thiazolindionové léčby.
Účinnost různých sil a dávek ezetimibu/simvastatinu (10/10 až 10/80 mg/den) byla prokázána v multicentrické, dvojitě zaslepené, placebem kontrolované 12týdenní studii s použitím všech dostupných dávek kombinace ezetimibu a simvastatinu a všech odpovídajících dávek simvastatinu. Při srovnání pacientů dostávajících všechny dávky kombinace ezetimibu a simvastatinu s pacienty, kteří dostávali všechny dávky simvastatinu, se ukázalo, že ezetimib/simvastatin významně snížil total-C, LDL-C a TG (viz Tabulka 1) jakož i Apo B (-42 % a – 29 %), non-HDL-C (-49 % a –34 %) a C-reaktivní protein (-33 % a –9 %, vždy v uvedeném pořadí). Účinky kombinace ezetimibu a simvastatinu na HDL-C se podobaly účinkům pozorovaným u simvastatinu. Další analýzy prokázaly, že ve srovnání s placebem ezetimib/simvastatin významně zvýšil koncentrace HDL-C.
| Léčba | |||||
| (denní dávka) | N | Total-C | LDL-C | HDL-C | TGa |
| Souhrnné údaje (všechny dávky ezetimibu/simvastatinu)c | 353 | –38 | –53 | +8 | –28 |
| Souhrnné údaje (všechny dávky simvastatinu)c | 349 | –26 | –38 | +8 | –15 |
| Ezetimib 10 mg | 92 | –14 | –20 | +7 | –13 |
| Placebo | 93 | +2 | +3 | +2 | –2 |
| ezetimib/simvastatin podle dávky | |||||
| 10/10 | 87 | –32 | –46 | +9 | –21 |
| 10/20 | 86 | –37 | –51 | +8 | –31 |
| 10/40 | 89 | –39 | –55 | +9 | –32 |
| 10/80 | 91 | –43 | –61 | +6 | –28 |
| Simvastatin podle dávky | |||||
| 10 mg | 81 | –21 | –31 | +5 | –4 |
| 20 mg | 90 | –24 | –35 | +6 | –14 |
| 40 mg | 91 | –29 | –42 | +8 | –19 |
| 80 mg | 87 | –32 | –46 | +11 | –26 |
a Pro triacylglyceroly střední % změna vůči výchozí
b Výchozí hodnota – ne při léčbě hypolipidemikem
c Souhrnně dávky ezetimibu/simvastatinu (10/10–10/80) ve srovnání se simvastatinem statisticky významně snížily celkový C, LDL-C a TG a ve srovnání s placebem statisticky významně zvýšily HDL-C.
Ve studii s podobným uspořádáním byly výsledky pro všechny lipidové parametry obecně shodné. V souhrnné analýze těchto dvou studií byla odpověď lipidů na ezetimib/simvastatin u pacientů s koncentracemi TG vyššími nebo nižšími než 200 mg/dl podobná.
V multicentrické, dvojitě zaslepené, kontrolované klinické studii (ENHANCE) bylo randomizováno 720 pacientů s heterozygotní familiární hypercholesterolemií do skupin léčených po dobu 2 let ezetimibem 10 mg v kombinaci se simvastatinem 80 mg (n = 357) nebo simvastatinem 80 mg (n = 363). Primárním cílem studie bylo zjištění účinku léčby kombinací ezetimib/simvastatin na tloušťku intimy medie (intima media thickness – IMT) karotidy v porovnání s monoterapií simvastatinem. Vliv zástupného markeru na kardiovaskulární morbiditu a mortalitu stále není prokázán.
Primární kritérium hodnocení, změna střední hodnoty IMT ve všech šesti segmentech karotidy, se mezi dvěma léčenými skupinami významně nelišilo (p = 0,29), jak bylo změřeno pomocí ultrazvuku v B modu. U ezetimibu 10 mg v kombinaci se simvastatinem 80 mg nebo u simvastatinu 80 mg samotného se za 2 roky trvání studie tloušťka intimy-medie zvýšila o 0,0111 mm, respektive o 0,0058 mm (výchozí střední hodnota IMT karotidy 0,68 mm, respektive 0,69 mm.
Ezetimib 10 mg v kombinaci se simvastatinem 80 mg snižoval LDL-C, celkový cholesterol, Apo B a TG významně více než simvastatin 80 mg. Procentuální vzestup HDL-C byl v obou léčených skupinách podobný. Nežádoucí účinky hlášené u ezetimibu 10 mg v kombinaci se simvastatinem 80 mg byly konzistentní s jeho známým bezpečnostním profilem.
Ezetimib/simvastatin obsahuje simvastatin. Ve dvou velkým placebem kontrolovaných klinických studiích, „Scandinavian Simvastatin Survival Study“ (20–40 mg; n = 4 444 pacientů) a „Heart Protection Study“ (40 mg; n = 20 536 pacientů), se hodnotily účinky podávání simvastatinu pacientům s vysokým rizikem koronárních příhod pro přítomnou ischemickou chorobu srdeční (ICHS), diabetes, ischemickou chorobu dolních končetin, anamnézu cévní mozkové příhody nebo jiného postižení mozkových cév. Prokázalo se, že simvastatin snížil: riziko celkové mortality snížením počtu úmrtí na ICHS; riziko nefatálního infarktu myokardu nebo cévní mozkové příhody a potřebu koronárních a nekoronárních revaskularizačních výkonů.
Studie „Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine“ (SEARCH) hodnotila účinky léčby simvastatinem v dávce 80 mg v porovnání s 20 mg (medián doby pozorování 6,7 roku) na velké cévní příhody (major vascular events MVEs; definované jako fatální ischemická choroba srdeční, nefatální infarkt myokardu, koronární revaskularizace, nefatální nebo fatální cévní mozková příhoda nebo periferní revaskularizace) u 12 064 pacientů s infarktem myokardu v anamnéze. V incidenci MVEs mezi těmito dvěma skupinami nebyl žádný významný rozdíl; simvastatin v dávce 20 mg (n = 1 553; 25,7 %) vs. simvastatin v dávce 80 mg (n = 1 477; 24,5 %); RR 0,94, 95 % interval spolehlivosti: 0,88 až 1,01. Absolutní rozdíl v LDL C mezi těmito dvěma skupinami byl v průběhu studie 0,35 ± 0,01 mmol/litr. Bezpečnostní profily byly v obou skupinách podobné s tou výjimkou, že incidence myopatie byla přibližně 1,0 % u pacientů léčených simvastatinem v dávce 80 mg v porovnání s 0,02 % u pacientů na dávce 20 mg. Přibližně polovina těchto případů myopatie se vyskytla během prvního roku léčby. Incidence myopatie během každého následujícího roku léčby byla přibližně 0,1 %.
Pediatrická populace
Klinické studie u pediatrických pacientů (10 až 17 let věku)
-
V multicentrické, dvojitě zaslepené, kontrolované studii bylo 142 chlapců (Tannerův stupeň II a vyšší) a 106 dívek po první menstruaci ve věku 10 až 17 let (střední hodnota věku 14,2 roku) s heterozygotní familiární hypercholesterolemií (HeFH) s výchozími hladinami LDL C mezi 4,1 a 10,4 mmol/l randomizováno buď do skupiny léčené ezetimibem 10 mg podávaným spolu se simvastatinem (10, 20 nebo 40 mg) nebo do skupiny léčené simvastatinem (10, 20 nebo 40 mg) samotným po dobu 6 týdnů, dalších 27 týdnů byl podáván ezetimib a 40 mg simvastatinu nebo samotného 40 mg simvastatinu a poté byl 20 týdnů v otevřeném uspořádání podáván ezetimib a simvastatin (10 mg, 20 mg nebo 40 mg).
-
V 6. týdnu ezetimib podávaný spolu se simvastatinem (všechny dávky) významně snižoval celkový cholesterol (38 % vs 26 %), LDL-C (49 % vs 34 %), Apo B (39 % vs 27 %) a non-HDL-C (47 % vs 33 %) v porovnání se simvastatinem samotným (všechny dávky). Výsledky v obou léčených skupinách byly podobné pro TG a HDL-C (-17 % vs –12 % a +7 % vs +6 %, v uvedeném pořadí). Ve 33. týdnu byly výsledky konzistentní s výsledky v 6. týdnu, přičemž významně více pacientů léčených ezetimibem a 40 mg simvastatinu (62 %) dosáhlo optimálních hodnot dle NCEP AAP (< 2,8 mmol/litr [110 mg/dl]) LDL C v porovnání s pacienty léčenými 40 mg simvastatinu (25 %). V 53. týdnu, na konci otevřeného prodloužení studie, byly účinky na lipidové parametry zachovány.
5.2 Farmakokinetické vlastnosti
Při podávání ezetimibu spolu se simvastatinem nebyly pozorovány žádné klinicky významné farmakokinetické interakce.
Absorpce
Ezetimib/simvastatin
Ezetimib/simvastatin představuje biologický ekvivalent současně podávanému ezetimibu a simvastatinu.
Ezetimib
Po perorálním podání se ezetimib rychle vstřebává a dochází k rozsáhlé konjugaci s farmakologicky aktivním fenolovým glukuronidem (ezetimib-glukuronid). Průměrných maximálních plazmatických koncentrací (Cmax) je dosaženo během 1 až 2 hodin u ezetimib-glukuronidu a 4 až 12 hodin u ezetimibu. Absolutní biologickou dostupnost ezetimibu nelze stanovit, protože uvedená látka je ve vodných médiích vhodných pro injekční podání prakticky nerozpustná.
Při perorálním ezetimibu ve formě 10mg tablet neměla současná konzumace jídla (s vysokým obsahem tuku a bez obsahu tuku) žádný vliv na biologickou dostupnost ezetimibu.
Simvastatin
Zjistilo se, že dostupnost účinné P-hydroxykyseliny v systémovém oběhu po perorálním podání jedné dávky simvastatinu je nižší než 5 % dávky, což je v souladu s rozsáhlým vylučováním po prvním průchodu játry. Hlavními metabolity simvastatinu přítomnými v lidské plazmě jsou P-hydroxykyselina a další čtyři účinné metabolity.
Ve srovnání se stavem nalačno nebyly plazmatické profily jak aktivních, tak i celkových inhibitorů ovlivněny, pokud se simvastatin podal okamžitě před zkušebním jídlem.
Distribuce
Ezetimib
Ezetimib se váže na proteiny lidské plazmy z 99,7 % a ezetimib-glukuronid z 88 až 92 %.
Simvastatin
Jak simvastatin, tak i P-hydroxykyselina se vážou na proteiny lidské plazmy (95 %).
Vyšetření farmakokinetiky simvastatinu po jednotlivých i opakovaných dávkách neprokázalo hromadění léčivé látky po opakovaných dávkách. Ve všech výše uvedených farmakokinetických studiích bylo maximální plazmatické koncentrace inhibitorů dosaženo 1,3 až 2,4 hodin po podání dávky.
Biotransformace
Ezetimib
Ezetimib je metabolizován převážně v tenkém střevě a v játrech cestou konjugace s glukuronidem (reakce II. fáze) s následným vyloučením žlučí. Minimální oxidační metabolizmus (reakce I. fáze) byl pozorován u všech hodnocených živočišných druhů. Hlavními látkami tvořícími se z léčivé látky a přítomnými v plazmě jsou ezetimib a ezetimib-glukuronid, představující přibližně 10 až 20 % a 80 až 90 % z celkového obsahu léčivé látky v plazmě (v uvedeném pořadí). Ezetimib i ezetimib-glukuronid se pozvolna vylučují z plazmy s prokazatelně významnou enterohepatální recyklací. Poločas ezetimibu i ezetimib-glukuronidu je přibližně 22 hodin.
Simvastatin
Simvastatin je neaktivní lakton, který se in vivo snadno rozkládá na příslušnou P-hydroxykyselinu, která je účinným inhibitorem HMG-CoA reduktázy. K hydrolýze dochází hlavně v játrech; rychlost hydrolýzy v lidské plazmě je velmi nízká.
U lidí se simvastatin dobře vstřebává a dochází k jeho rozsáhlému vylučování při prvním průchodu játry. Vylučování játry závisí na průtoku krve játry. Játra jsou hlavním místem účinku s následným vylučováním ekvivalentů jednotlivých léčivých látek do žluči. Dostupnost účinné látky v systémovém oběhu je proto malá.
Po nitrožilní aplikaci P-hydroxykyseliny jako metabolitu dosahoval její poločas v průměru 1,9 hodiny.
Eliminace
Ezetimib
Po perorálním podání 14C-ezetimibu (20 mg) lidem představoval celkový ezetimib přibližně 93 % celkové radioaktivity v plazmě. Přibližně 78 % aplikované radioaktivity bylo v průběhu 10denního sběrného období zjištěno ve stolici a 11 % v moči. Po 48 hodinách nebyly v plazmě přítomny zjistitelné koncentrace radioaktivity.
Simvastatin
Simvastatin je aktivně vychytáván do hepatocytů transportérem OATP1B1.
Simvastatin je substrát efluxního transportéru BCRP.
Po perorálním podání dávky radioaktivního simvastatinu lidem se do 96 hodin 13 % radioaktivity vyloučilo močí a 60 % stolicí. Množství zjištěné ve stolici představuje vstřebané ekvivalenty léčivé látky vyloučené žlučí i nevstřebané látky. Po nitrožilní aplikaci metabolitu P-hydroxykyseliny se v průměru pouze 0,3 % i.v. dávky vyloučily močí jako inhibitory.
Zvláštní skupiny pacientů
Pediatrická populace
Vstřebávání a metabolizmus ezetimibu u dětí, dospívajících (10 až 18 let) a dospělých jsou si podobné. Na základě celkového ezetimibu, neexistují mezi dospívajícími a dospělými rozdíly ve farmakokinetice. Farmakokinetické údaje pro dětskou populaci ve věku < 10 let nejsou k dispozici. Zkušenosti z klinické praxe s dětmi a dospívajícími jsou omezeny na pacienty s HoFH, HeFH nebo sitosterolemií (viz bod 4.2.)
Starší pacienti
Plazmatická koncentrace celkového ezetimibu je u starších jedinců (> 65 let) přibližně dvakrát vyšší než u mladších jedinců (18 až 45 let). Snížení koncentrací LDL-C a profil bezpečnosti u starších a mladších jedinců léčených ezetimibem jsou srovnatelné. (Viz bod 4.2.)
Pacienti s poruchou funkce jater
Po jednorázové 10mg dávce ezetimibu se průměrná hodnota AUC celkového ezetimibu u pacientů s mírnou poruchou funkce jater (Child-Pughovo skóre 5 nebo 6) ve srovnání se zdravými jedinci zvětšila přibližně 1,7násobně. Ve 14denní studii s opakovanými dávkami (10 mg denně) podávanými pacientům se středně těžkou poruchou funkce jater (Child-Pughovo skóre 7 až 9) se průměrná hodnota AUC celkového ezetimibu zvýšila ve srovnání se zdravými jedinci v den 1 a v den 14 přibližně 4násobně. U pacientů s lehkou poruchou funkce jater není nutno dávku nijak upravovat. Vzhledem k tomu, že účinky zvýšené expozice ezetimibu u pacientů se středně těžkou nebo těžkou (Child-Pughovo skóre > 9) poruchou funkce jater nejsou známy, nedoporučuje se ezetimib u uvedených skupin pacientů používat (viz body 4.2 a 4.4).
Pacienti s poruchou funkce ledvin
Ezetimib
Po jednorázové 10mg dávce ezetimibu pacientům s těžkým onemocněním ledvin (n = 8; průměrná CrCl < 30 ml/min) se ve srovnání se zdravými jedinci (n = 9) průměrná hodnota AUC celkového ezetimibu zvětšila přibližně 1,5násobně. (Viz bod 4.2.)
U dalšího pacienta v uvedené studii (po transplantaci ledvin a užívající několik léčiv včetně cyklosporinu) byla zjištěna 12násobně zvýšená expozice celkovému ezetimibu.
Simvastatin
Ve studii s pacienty s těžkou poruchou funkce ledvin (clearance kreatininu < 30 ml/min) byly plazmatické koncentrace celkových inhibitorů po jednorázové dávce příbuzného inhibitoru HMG-CoA reduktázy přibližně dvakrát vyšší než u zdravých dobrovolníků.
Pohlaví
Plazmatické koncentrace celkového ezetimibu jsou u žen mírně vyšší (přibližně o 20 %) než u mužů. U mužů i u žen léčených ezetimibem jsou snížení koncentrace LDL-C a profil bezpečnosti srovnatelné.
Polymorfismus SLCO1B1
Nosiči genu SLCO1B1, alely c.521T>C, mají nižší aktivitu OATP1B1. Průměrná expozice (AUC) hlavnímu aktivnímu metabolitu kyseliny simvastatinové je 120 % u heterozygotních nosičů alely C (CT) a 221 % u homozygotních nosičů (CC) v porovnání s pacienty, kteří mají nejčastěji se vyskytující genotyp (TT). Alela C má v evropské populaci frekvenci výskytu 18 %. U pacientů s polymorfismem SLCO1B1 je riziko zvýšené expozice simvastatinu, což může vést ke zvýšenému riziku rhabdomyolýzy (viz bod 4.4).
5.3 Preklinické údaje vztahující se k bezpečnosti
Ezetimib/simvastatin
Ve studiích se současným podáváním ezetimibu a simvastatinu byly pozorovány toxické účinky v podstatě takové, jaké se typicky vyskytují v souvislosti s podáváním statinů. Některé z toxických účinků byly výraznější než během léčby samotnými statiny. Tato skutečnost se připisuje farmakokinetickým a/nebo farmakodynamickým interakcím po současném podání obou léčivých látek. V klinických studiích se takové interakce nevyskytly. Myopatie se vyskytly u potkanů až po expozici dávkám několikanásobně vyšším než je terapeutická dávka u člověka (přibližně 20násobek velikosti AUC pro simvastatin a 1 800násobek velikosti AUC pro účinný metabolit). Neexistují žádné důkazy, že by současné podávání ezetimibu ovlivnilo myotoxický potenciál simvastatinu.
U psů, jimž byl současně podáván ezetimib a statiny, byly v nízkých expozicích (< 1krát AUC u člověka) pozorovány některé účinky na játra. Byla pozorována výrazná zvýšení jaterních enzymů (ALT, AST) bez přítomnosti tkáňových nekróz. Histopatologické nálezy na játrech (hyperplazie žlučových cest, akumulace pigmentu, infiltrace mononukleárních buněk a malé hepatocyty) byly pozorovány u psů, jimž byl podáván současně ezetimib a simvastatin. Tyto změny se delším podáváním až do 14 měsíců nezvětšovaly. Celková obnova jaterních nálezů byla pozorována po přerušení dávek. Tyto nálezy byly shodné s těmi, jež byly popsány u inhibitorů HMG-CoA nebo jsou přisuzovány velmi nízkým hladinám cholesterolu dosaženým u postižených psů.
Současné podávání ezetimibu a simvastatinu nemělo u potkanů teratogenní účinky. U březích samic králíků byl pozorován malý počet kosterních deformit (srůst kaudálních obratlů, snížený počet kaudálních obratlů).
V řadě in vivo a in vitro studií nevykazoval ezetimib, ať již podávaný samostatně nebo spolu se simvastatinem, žádný genotoxický potenciál.
Ezetimib
Studie chronické toxicity ezetimibu u zvířat neprokázaly žádný cílový orgán pro toxické účinky. U psů, jimž byl po dobu čtyř týdnů podáván ezetimib (> 0,03 mg/kg/den) se koncentrace cholesterolu ve žlučníkové žluči zvýšily 2,5 až 3,5násobně. V jednoleté studii u psů, jimž byly podávány dávky až 300 mg/kg/den však nebyly pozorovány zvýšená incidence cholelitiázy ani jiné hepatobiliární účinky. Význam těchto zjištění pro člověka není znám. Litogenní riziko při léčebném používání ezetimibu nelze vyloučit.
Dlouhodobé zkoušky kancerogenity ezetimibu byly negativní.
Ezetimib nemá žádný účinek na plodnost samců ani samic potkanů, ani nebyla prokázána jeho teratogenita u potkanů a králíků, neměl vliv ani na prenatální a postnatální vývoj. U březích samic potkanů a králíků, jimž byly opakovaně podávány dávky 1 000 mg/kg/den prostupoval ezetimib placentární bariérou.
Simvastatin
Podle konvenčních studií se zvířaty zaměřených na farmakodynamiku, toxicitu opakovaných dávek, genotoxicitu a kancerogenitu neexistují pro pacienty žádná rizika, která by bylo možno očekávat na základě farmakologického mechanismu. Při maximálních tolerovaných dávkách u potkanů a králíků nevedlo podávání simvastatinu ke vzniku malformací u plodu a neměl žádný vliv na plodnost, funkci reprodukce ani vývoj novorozenců.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Monohydrát laktózy
Monohydrát kyseliny citronové Butylhydroxyanisol (E320)
Kyselina askorbová (E300)
Natrium-lauryl-sulfát (E487)
Sodná sůl kroskarmelózy
Hypromelóza (E464)
Mikrokrystalická celulóza
Magnesium- stearát
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
Pouze lahvičky: Spotřebujte do 100 dní od otevření
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
PVC/Aclar-Al blistry:
10 mg/10 mg: 14, 28, 30 a 100 tablet
10 mg/20 mg: 14, 28, 30, 98, 100 tablet a kalendářní balení obsahující 28 tablet
Perforované jednodávkové PVC/Aclar-Al blistry:
10 mg/10 mg: 14 × 1 a 28 × 1 tableta
10 mg/20 mg: 14 × 1, 28 × 1, 30 × 1 a 90 × 1 tableta
Plastové HDPE lahvičky se šroubovacím polypropylenovým (PP) uzávěrem, těsněním z hliníkové folie a absorpční vatou:
10 mg/10 mg: 30 a 100 tablet
10 mg/20 mg: 30, 100, 250 a 500 tablet
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Mylan Ireland Limited
Unit 35/36 Grange Parade
Baldoyle Industrial Estate
Dublin 13
Irsko
8. REGISTRAČNÍ ČÍSLO(A)
Ezetimib/Simvastatin Mylan 10 mg/10 mg: 31/271/15-C
Ezetimib/Simvastatin Mylan 10 mg/20 mg: 31/272/15-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 3. 6. 2015
Další informace o léčivu EZETIMIB/SIMVASTATIN MYLAN
Jak
se EZETIMIB/SIMVASTATIN MYLAN
podává: perorální podání - tableta
Výdej
léku: na lékařský předpis
Balení: Blistr
Velikost
balení: 28 KALBAL
Držitel rozhodnutí o registraci daného léku v České republice:
Generics [UK] Limited, Potters Bar