Souhrnné informace o léku - ELIGARD
1. NÁZEV PŘÍPRAVKU
ELIGARD 22,5 mg prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s práškem pro injekční roztok obsahuje leuprorelini acetas
-
22,5 mg, což odpovídá leuprorelinum 20,87 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek (stříkačka B):
Předplněná stříkačka obsahující bílý až téměř bílý prášek.
Rozpouštědlo (stříkačka A):
Předplněná stříkačka obsahující čirý, bezbarvý až bledě žlutý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
ELIGARD 22,5 mg je indikován k léčbě pokročilého hormonálně dependentního karcinomu prostaty a k léčbě vysoce rizikového, lokalizovaného a lokálně pokročilého hormonálně dependentního karcinomu prostaty v kombinaci s radioterapií.
4.2 Dávkování a způsob podání
Dávkování
Dospělí muži
ELIGARD 22,5 mg má být podáván pod dohledem lékaře s odbornými znalostmi potřebnými pro sledování odpovědi na léčbu.
ELIGARD 22,5 mg je podáván jako jednorázová podkožní injekce jednou za 3 měsíce. Injektovaný roztok vytvoří depo léčiva, které průběžně uvolňuje leuprorelin-acetát po dobu 3 měsíců.
Léčba pokročilého karcinomu prostaty přípravkem ELIGARD 22,5 mg je v zásadě dlouhodobá. Terapie se nemá přerušovat, dojde-li k remisi nebo zlepšení.
ELIGARD 22,5 mg má být používán jako neoadjuvantní nebo adjuvantní terapie v kombinaci s radioterapií u vysoce rizikového lokalizovaného a lokálně pokročilého karcinomu prostaty.
Odezva na ELIGARD 22,5 mg se musí sledovat pomocí klinických parametrů a měřením hladin prostatického specifického antigenu (PSA) v séru. Klinické studie ukázaly, že se hladina testosteronu u většiny nekastrovaných pacientů během prvních 3 dnů zvýšila a poté se během 3 –4 týdnů snížila pod kastrační úroveň. Po dosažení této hladiny se kastrační úroveň udržovala po celou dobu trvání terapie (< 1,0 % výkyvů v hladině testosteronu). Pokud není odezva pacienta zcela optimální, je vhodné ověřit, zda hladiny testosteronu v séru dosáhly kastrační úrovně a setrvávají na ní. Vzhledem k tomu, že v důsledku nesprávné přípravy, rekonstituce nebo podávání přípravku může dojít k nedostatečnému klinickému účinku, musí být v případě nesprávného zacházení nebo podezření na ně vyhodnoceny hladiny testosteronu (viz bod 4.4).
U pacientů s metastatickým kastračně resistentním karcinomem prostaty léčených agonisty GnRH, jako je leuprorelin, u kterých nebyla provedena chirurgická kastrace, a jsou způsobilí pro léčbu inhibitory androgenní biosyntézy nebo inhibitory androgenních receptorů, lze pokračovat v léčbě agonisty GnRH.
Pediatrická populace
Bezpečnost a účinnost u dětí ve věku 0 až 18 let nebyla stanovena (viz též bod 4.3).
Zvláštní skupiny pacientů
Nebyly provedeny žádné klinické studie s pacienty s poruchou funkce jater nebo ledvin.
Způsob podání
Přípravek ELIGARD 22,5 mg musí být připravován, rekonstituován a podáván pouze zdravotnickým pracovníkem, který je seznámen s těmito postupy. Viz bod 6.6:_Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním. Pokud není přípravek připraven správně, nesmí být podáván.
Obsah dvou předplněných sterilních injekčních stříkaček se musí smísit bezprostředně před subkutánní aplikací přípravku ELIGARD 22,5 mg.
Na základě údajů ze zkoušek na zvířatech je naprosto nezbytné se vyvarovat intraarteriální nebo intravenózní aplikace.
Stejně jako u jiných léčivých přípravků podávaných subkutánně má být místo vpichu pravidelně obměňováno.
4.3 Kontraindikace
ELIGARD 22,5 mg je kontraindikován u žen a dětí.
Přecitlivělost na leuprorelin-acetát, jiné agonisty GnRH nebo kteroukoli z pomocných látek přípravku uvedených v bodě 6.1.
U pacientů po orchiektomii (v případě chirurgické kastrace nemá ELIGARD 22,5 mg, stejně jako ostatní agonisté GnRH, za následek další snížení testosteronu v séru).
Podávání přípravku v monoterapii u pacientů s karcinomem prostaty a současně s kompresí míchy nebo prokázanými míšními metastázami (též viz bod 4.4).
4.4 Zvláštní upozornění a zvláštní opatření pro použití
Správná rekonstituce: V důsledku nesprávné rekonstituce přípravku může dojít k nedostatečnému klinickému účinku. Popis postupu přípravy a podávání přípravku a vyhodnocení hladin testosteronu v případě nesprávného zacházení nebo podezření na ně viz bod 4.2 a bod 6.6.
Androgen-deprivační léčba může prodlužovat QT interval:
Před zahájením léčby přípravkem ELIGARD 22,5 mg má lékař zvážit poměr přínosů a rizik, včetně rizika torsade de pointes, u pacientů s rizikovými faktory pro prodloužení QT intervalu v anamnéze a u pacientů souběžně užívajících léčivé přípravky, které mohou prodlužovat QT interval (viz bod 4.5).
Kardiovaskulární onemocnění: V souvislosti s používáním agonistů GnRH u mužů bylo hlášeno zvýšené riziko vzniku infarktu myokardu, náhlé srdeční smrti a cévní mozkové příhody. Na základě hlášené míry pravděpodobnosti se riziko zdá nízké, má být při stanovení léčby pacientů s karcinomem prostaty pečlivě vyhodnoceno společně s kardiovaskulárními rizikovými faktory. Pacienti, kteří jsou léčeni agonisty GnRH, mají být monitorováni s ohledem na příznaky a známky rozvoje kardiovaskulárního onemocnění a léčeni podle současné klinické praxe.
Přechodné zvýšení hladiny testosteronu: Leuprorelin-acetát, stejně jako ostatní agonisté GnRH, způsobuje během prvních týdnů léčby přechodné zvýšení koncentrací testosteronu, dihydrotestosteronu a kyselé fosfatázy v séru. Pacienti mohou pozorovat zhoršení subjektivních příznaků nebo výskyt nových příznaků, včetně bolestí v kostech, neuropatie, hematurie nebo obstrukce močových cest nebo močového měchýře (viz bod 4.8). Tyto příznaky obvykle v průběhu terapie ustupují.
Je třeba zvážit doplňkové podání vhodného antiandrogenu, a to 3 dny před zahájením léčby leuprorelinem a dále v průběhu prvních 2 až 3 týdnů léčby. Bylo prokázáno, že toto opatření chrání před následky počátečního zvýšení sérové hladiny testosteronu.
Podávání přípravku ELIGARD 22,5 mg po chirurgické kastraci již nevede k dalšímu snížení hladin testosteronu v séru u mužů.
Denzita kostí: U mužů po kastraci nebo u mužů, kteří byli léčeni agonisty GnRH (viz bod 4.8), bylo v literatuře popsáno řídnutí kostí.
Terapie antiandrogeny významně zvyšuje riziko fraktur z důvodu osteoporózy. K tomuto problému existuje zatím málo informací. Fraktury z důvodu osteoporózy byly pozorovány u 5 % pacientů po 22 měsících terapie založené na farmakologické deprivaci androgenu a u 4 % pacientů po 5 až 10 letech této léčby. Riziko fraktur z důvodu osteoporózy je obecně vyšší než riziko patologických fraktur.
Na rozvoji osteoporózy se kromě dlouhodobého deficitu testosteronu mohou podílet také vyšší věk, kouření a požívání alkoholických nápojů, obezita a nedostatek pohybu.
Hypofyzární apoplexie: Během postmarketingového sledování byly po podání GnRH agonistů zaznamenány vzácné případy hypofyzární apoplexie (klinický syndrom související s infarktem hypofýzy). Většina příznaků se objevila během 2 týdnů po podání první dávky, některé během první hodiny. V těchto případech se hypofyzární apoplexie projevovala náhlou bolestí hlavy, zvracením, změnami vidění, oftalmoplegií, změnami psychického stavu a někdy kardiovaskulárním kolapsem. Stav vyžaduje okamžitý lékařský zásah.
Hyperglykemie a diabetes: Hyperglykemie a zvýšené riziko vzniku diabetu byly hlášeny u mužů, kterým jsou podávány agonisté GnRH. Hyperglykemie může znamenat rozvoj diabetu mellitu nebo zhoršení kontroly glykemie u pacientů s diabetem. U pacientů, kterým jsou pravidelně podávány agonisté GnRH, mají být pravidelně měřeny hladiny glukózy a/nebo glykosylovaného hemoglobinu (HbA1c) v krvi a má se postupovat podle současné praxe při léčbě hyperglykemie nebo diabetu.
Křeče: Z postmarketingových hlášení byl pozorován výskyt křečí u pacientů, kterým je podáván leuprolerin-acetát a u kterých se predispoziční faktory v minulosti vyskytovaly i nevyskytovaly. Při výskytu křečí je třeba se řídit relevantními doporučeními současné klinické praxe.
Ostatní příhody: Případy obstrukce močových cest a komprese míchy, které se mohou podílet na paralýze s fatálními komplikacemi nebo bez nich, byly pozorovány při podávání agonistů GnRH. Pokud dojde ke vzniku komprese míchy nebo poruchy funkce ledvin, je nutno zahájit standardní léčbu těchto komplikací.
Pacienti s vertebrálními nebo mozkovými metastázami a pacienti s obstrukcí močových cest musí být během prvních týdnů léčby pozorně sledováni.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Pro přípravek ELIGARD 22,5 mg nebyly provedeny žádné farmakokinetické studie lékových interakcí. Nejsou rovněž k dispozici žádné zprávy o jakýchkoli interakcích leuprorelin-acetátu s jinými léčivými přípravky.
Kvůli souvislosti androgen-deprivační léčby a prodloužení QT intervalu má být pečlivě zvážena souběžná léčba přípravkem ELIGARD 22,5 mg s léčivými přípravky, o kterých je známo, že prodlužují QT interval, a léčba přípravky, které mohou vyvolat torsade de pointes, jako antiarytmika třídy I A (např. chinidin, disopyramid), třídy III (např. amiodaron, sotalol, dofetilid, ibutilid), methadon, moxifloxacin, antipsychotika a další (viz bod 4.4.).
4.6 Fertilita, těhotenství a kojení
Neuplatňuje se vzhledem k tomu, že ELIGARD 22,5 mg je u žen kontraindikován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie účinků přípravku ELIGARD® 22,5 mg na schopnost řídit a obsluhovat stroje.
Schopnost řídit a obsluhovat stroje může být ovlivněna v důsledku únavy, závratí a poruch vidění jako možných nežádoucích účinků léčby nebo projevů základního onemocnění.
4.8 Nežádoucí účinky
Nežádoucí účinky pozorované u přípravku ELIGARD se týkají zejména specifické farmakologické aktivity leuprorelin-acetátu, jmenovitě zvyšování a snižování hladin určitých hormonů. Nejčastěji hlášenými nežádoucími účinky jsou návaly horka, nauzea, malátnost a únava a přechodné místní podráždění v místě vpichu. Mírné návaly horka se vyskytují u 58 % pacientů.
Tabulkový přehled nežádoucích účinků
Následující nežádoucí účinky byly hlášeny během klinických studií s přípravkem ELIGARD u pacientů s pokročilým karcinomem prostaty. Tyto nežádoucí účinky jsou klasifikovány podle četnosti výskytu jako velmi časté (>1/10), časté (>1/100, <1/10), méně časté (>1/1 000, <1/100), vzácné (>1/10 000, <1/1 000) a velmi vzácné (<1/10 000), není známo (nelze ji z dostupných údajů určit).
| Tabulka 1: Nežádoucí účinky v klinických studiích s přípravkem ELIGARD | |
| Infekce a infestace časté méně časté | nazofaryngitida infekce močových cest, lokální kožní infekce |
| Poruchy metabolismu a výživy méně časté | zhoršení diabetes mellitus |
| Psychiatrické poruchy méně časté | neobvyklé sny, deprese, snížení libida |
| Poruchy nervového systému méně časté vzácné | závratě, bolest hlavy, hypestezie, nespavost, poruchy chuti a čichu, vertigo abnormální bezděčné pohyby |
| Srdeční poruchy není známo | prodloužení QT intervalu (viz bod 4.4 a 4.5) |
| Cévní poruchy velmi časté méně časté vzácné | návaly horka hypertenze, hypotenze synkopa, kolaps |
| Respirační, hrudní a mediastinální poruchy méně časté není známo | výtok z nosu, dušnost intersticiální plicní onemocnění |
| Gastrointestinální poruchy časté méně časté vzácné | nauzea, průjem, gastroenteritida/kolitida zácpa, sucho v ústech, dyspepsie, zvracení nadýmání, říhání |
| Poruchy kůže a podkožní tkáně velmi časté časté méně časté vzácné | ekchymózy, erytém svědění, noční pocení, lepkavost kůže, nadměrné pocení alopecie, kožní výsevy |
| Poruchy svalové a kosterní soustavy a pojivové tkáně časté méně časté | bolest kloubů, bolest končetin, bolest svalů, třesavky, slabost bolest zad, svalové křeče |
| Tabulka 1: Nežádoucí účinky v klinických studiích s přípravkem ELIGARD | |
| Poruchy ledvin a močových cest časté méně časté | snížená frekvence močení, obtíže při močení, dysurie, nykturie, oligurie spasmus měchýře, hematurie, časté nucení na močení, retence moči |
| Poruchy reprodukčního systému a prsu časté méně časté vzácné | zvýšená citlivost prsů, atrofie varlat, bolest varlat, neplodnost, hypertrofie prsů, erektilní dysfunkce, zmenšení velikosti penisu gynekomastie, impotence, poruchy varlat bolest prsů |
| Celkové poruchy a reakce v místě aplikace velmi časté časté méně časté vzácné velmi vzácné | únava, pálení v místě vpichu, mravenčení v místě vpichu malátnost, bolest místa vpichu, zhmožděniny místa vpichu, štípání v místě vpichu svědění místa vpichu, indurace místa vpichu, netečnost, bolest, pyrexie ulcerace místa vpichu nekróza místa vpichu |
| Poruchy krve a lymfatického systému časté | hematologické změny, anémie |
| Vyšetření časté méně časté | zvýšená hladina kreatininfosfokinázy v krvi, prodloužený čas koagulace zvýšení hladiny alaninaminotransferázy, zvýšená hladina triglyceridů v krvi, prodloužený protrombinový čas, zvýšení tělesné hmotnosti |
K dalším nežádoucím účinkům všeobecně hlášeným při terapii leuprorelin-acetátem patří periferní otok, plicní embolie, palpitace, myalgie, svalová slabost, změny kožního čití, zimnice, vyrážka, amnézie a poruchy vidění. U dlouhodobého užívání přípravků této skupiny byla pozorována muskulární atrofie. Po podání krátkodobě i dlouhodobě působících agonistů GnRH byl vzácně zaznamenán infarkt v místě hypofyzární apoplexie. Vzácně byly hlášeny případy trombocytopenie a leukopenie. Byly popsány změny glukózové tolerance.
Po podání analogů agonistů GnRH byly hlášeny křeče (viz bod 4.4).
Lokální nežádoucí příhody po injekci přípravku ELIGARD jsou podobné lokálním nežádoucím příhodám vyskytujícím se u podobných subkutánně podávaných přípravků.
Obecně lze říci, že tyto lokální nežádoucí účinky vzniklé po podkožní injekci jsou mírné a mají krátké trvání.
Anyfylaktické/anafylaktoidní reakce byly hlášeny vzácně po podání analogů agonistů GnRH.
Změny v hustotě kostí
O snížené hustotě kostí bylo v lékařské literatuře referováno v případě mužů, kteří podstoupili kastraci nebo byli léčeni analogy GnRH. Lze předpokládat, že dlouhodobá léčba leuprorelinem může vykazovat nárůst příznaků osteoporózy. Podrobnosti ke zvýšenému riziku zlomenin z osteoporózy (viz bod 4.4).
Zhoršení příznaků a projevů onemocnění
Léčba leuprorelin-acetátem může způsobit zhoršení příznaků a projevů onemocnění během prvních několika týdnů. Pokud dojde ke zhoršení podmínek jako např. vertebrálních metastáz a/nebo obstrukce močových cest nebo hematurie, mohou nastat neurologické obtíže, jako je slabost a/nebo parestézie dolních končetin nebo zhoršení příznaků týkajících se močových cest.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
ELIGARD 22,5 mg nemá potenciál k abúzu a náhodné předávkování je nepravděpodobné. V klinické praxi nebyly zaznamenány žádné případy abúzu nebo předávkování leuprorelin-acetátem. Pokud k nadměrné expozici přípravku dojde, doporučuje se sledování pacienta a podpůrná symptomatická léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Analogy gonadotropin-releasing hormonu
ATC kód: L02A E02
Leuprorelin-acetát je syntetický nonapeptidový agonista přirozeně se vyskytujícího gonadoliberinu (GnRH), který, pokud se podává trvale, inhibuje hypofyzární sekreci gonadotropinu a potlačuje tvorbu steroidů ve varlatech u mužů. Tento účinek je po přerušení terapie reverzibilní. Agonista je nicméně účinnější než přirozený hormon a doba do návratu hladiny testosteronu na původní hodnotu se může u jednotlivých pacientů lišit.
Podávání leuprorelin-acetátu má za následek počáteční zvýšení hladin cirkulujícího luteinizačního hormonu (LH) a folikulostimulačního hormonu (FSH), což vede k přechodnému zvýšení hladin gonadálních steroidů, testosteronu a dihydrotestosteronu u mužů. Trvalé podávání leuprorelin-acetátu vede ke snížení hladin LH a FSH. U mužů se hladina testosteronu snižuje pod úroveň kastračního prahu (< 50 ng/dl). K těmto poklesům dochází během tří až pěti týdnů po zahájení léčby. Průměrné hladiny testosteronu po šesti měsících jsou 10,1 (±0,7) ng/dl, což je srovnatelné s hladinami po oboustranné kastraci. Všichni pacienti, kteří v pivotní klinické studii dostávali plnou dávku 22,5 mg leuprorelinu dosáhli kastračních hladin do 5 týdnů; 99 % do 28. dne. U velké většiny pacientů byly pozorovány hodnoty testosteronu pod 20 ng/dl, ačkoliv plný pozitivní dopad těchto nízkých hladin se ještě neprojevil. Hladiny PSA po šesti měsících poklesly o 98%.
Dlouhodobé studie ukázaly, že pokračování terapie udržuje hladinu testosteronu pod kastračním prahem po dobu až sedmi let a pravděpodobně již navždy.
V průběhu klinického zkoušení nebyla přímo měřena velikost tumoru, při léčbě přípravkem ELIGARD 22,5 mg však byla pozorována příznivá odezva ve smyslu snížení průměrné hladiny PSA o 98 %.
Ve fázi III randomizované klinické studie, které se zúčastnilo 970 pacientů s lokálně pokročilým karcinomem prostaty (hlavně pacienti T2c-T4 a někteří T1c až T2b s onemocněním regionálních lymfatických uzlin), ze kterých bylo 483 zařazeno do skupiny s krátkodobou androgenní supresí (6 měsíců) v kombinaci s radioterapií a 487 do skupiny s dlouhodobou terapií (3 roky), byla při non-inferiorní analýze porovnávána krátkodobá a dlouhodobá konkomitantní a adjuvantní hormonální léčba GnRH agonisty (triptorelinem nebo goserelinem). Celková 5letá mortalita byla 19% ve skupině s krátkodobou léčbou a 15,2 % s dlouhodobou léčbou. Zaznamenaný poměr rizik 1,42 s horním jednostranným 95,71 % CI 1,79 nebo dvoustranným 95,71% CI 1,09; 1,85 (p = 0,65 pro průkaz non-inferiority) demonstruje, že kombinace radioterapie s 6měsíční androgen deprivační terapií vede k horšímu přežití ve srovnání s radioterapií spolu s 3letou androgen deprivační terapií. Celkové 5leté přežití ve skupině s dlouhodobou a krátkodobou léčbou dosahuje 84,8 % a 81,0 %. Celková kvalita života při použití QLQ-C30 se statisticky významně neliší mezi těmito dvěma skupinami (P=0,37). Ve výsledcích převládají data populace pacientů s lokálně pokročilým nádorem.
Důkazy pro indikaci vysoce rizikového lokalizovaného karcinomu prostaty vycházejí z publikovaných studií radioterapie v kombinaci s analogy GnRH, včetně leuprorelin-acetátu. Byla analyzována data z pěti publikovaných klinických studií (EORTC 22863, RTOG 85–31, RTOG 92–02, RTOG 8610, a D'Amico et al., JAMA, 2004), která všechna demonstrují přínos kombinace analogů GnRH s radioterapií. Jednoznačné rozlišení cílové populace pro indikaci lokálně pokročilého karcinomu prostaty a vysoce rizikového lokalizovaného karcinomu prostaty nebylo v publikovaných studiích možné.
Klinická data ukazují, že radioterapie s následnou 3letou androgen deprivační léčbou je vhodnější než radioterapie s následnou 6měsíční androgen deprivační terapií.
Doporučená délka androgen deprivační terapie v léčebných doporučeních pro pacienty klinického stadia T3 – T4, kteří podstupují radioterapii, je 2 – 3 roky.
5.2 Farmakokinetické vlastnosti
Absorpce: U pacientů s pokročilým karcinomem prostaty po počáteční injekci prudce vzrostou průměrné koncentrace leuprorelinu v séru na 127 ng/ml 4,6 hod (Cmax) po injekci. Po počátečním zvýšení koncentrace v séru po každé injekci (fáze plató 3 – 84 dnů po každé dávce) zůstávají koncentrace v séru relativně konstantní (0,2 – 2 ng/ml). Neexistují žádné známky akumulace po opakovaném podávání.
Distribuce: Střední ustálený distribuční objem leuprorelinu po podání intravenózního bolusu zdravým mužským dobrovolníkům byl 27 litrů. Míra vazby na lidské plazmatické proteiny in vitro se pohybovala od 43 % do 49 %.
Eliminace: U zdravých mužských dobrovolníků vykázal 1 mg leuprorelin-acetátu podaný jako intravenózní bolus střední systémovou clearance 8,34 l/h, s poločasem eliminace zhruba 3 hodiny (na základě dvojkompartmentového modelu).
S přípravkem ELIGARD nebyly provedeny žádné studie exkrece.
S přípravkem ELIGARD nebyly provedeny žádné studie metabolizace.
5.3 Předklinické údaje vztahující se k bezpečnosti
Účinky na reprodukční systém, které byly odhaleny v preklinických studiích s leuprorelin-acetátem prováděných na obou pohlavích, odpovídaly známým farmakologickým vlastnostem této látky. Tyto účinky se ukázaly být reverzibilní po vysazení léčby a příslušném období regenerace. Leuprorelin-acetát nevykazoval teratogenitu. U králíků byla pozorována embryotoxicita/letalita v souladu s farmakologickými účinky leuprorelin-acetátu na reprodukční systém.
Studie karcinogenity v délce 24 měsíců byly provedeny na potkanech a myších. Po podkožním podání bylo u potkanů pozorováno na dávkách závislé zvýšení frekvence apoplexií hypofýzy, a to při dávkách 0,6 až 4 mg/kg/den. Žádný takový účinek nebyl pozorován u myší.
V sérii in vitro a in vivo zkoušek nevykazoval leuprorelin-acetát ani odpovídající přípravek ELIGARD 22,5 mg (s uvolňováním po dobu 1 měsíce) mutagenní potenciál.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Rozpouštědlo (injekční stříkačka A): polyglaktin (1:3),methylpyrrolidon
Prášek (injekční stříkačka B): žádné
6.2 Inkompatibility
Leuprorelin obsažený v injekční stříkačce B může být smísen pouze s rozpouštědlem v injekční stříkačce A a nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky
Jakmile byl přípravek vyjmut mimo chladničku, smí být uchováván v původním obalu při pokojové teplotě (do 25 °C) maximálně po dobu čtyř týdnů.
Po prvním otevření vaničky musí být prášek a rozpouštědlo pro injekční roztok okamžitě smíseny a podány pacientovi.
Po rekonstituci ihned aplikujte, protože s přibývajícím časem se zvyšuje viskozita roztoku.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C – 8 °C) v původním obalu, aby byl přípravek chráněn před vlhkostí.
Přípravek musí mít před podáním pokojovou teplotu. Vyjměte přípravek z chladničky přibližně 30 minut před podáním. Jakmile je přípravek mimo chladničku, smí být uchováván v původním obalu při pokojové teplotě (do 25 °C) maximálně po dobu čtyř týdnů.
6.5 Druh obalu a obsah balení
Dvě injekční stříkačky: jedna injekční stříkačka vyrobená z cykloolefínového kopolymeru obsahující prášek (stříkačka B) a jedna injekční stříkačka vyrobená z polypropylenu obsahující rozpouštědlo (stříkačka A). Obě stříkačky spolu tvoří systém pro smísení.
Injekční stříkačka A má zakončení pístu z termoplastické pryže a je uzavřena polyethylenovým nebo polypropylenovým Luer-Lock krytem. Zátka a oba písty injekční stříkačky B jsou vyrobeny z chlorbutylové pryže.
Přípravek je dostupný v následujících velikostech balení:
- Souprava obsahující dvě zatavené vaničky v papírové krabičce. Jedna vanička obsahuje předplněnou polypropylenovou injekční stříkačku A, tyčinku s dlouhým pístem a sáček s vysoušedlem. Druhá vanička obsahuje předplněnou injekční stříkačku B vyrobenou z cykloolefínového kopolymeru, sterilní jehlu velikosti 20 G a sáček se silikonovým vysoušedlem.
- Sada obsahující soupravy sestávající z 2 × 2 injekčních stříkaček (1 x stříkačka A; 1 x stříkačka B).
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Nechte přípravek ohřát na pokojovou teplotu, vyjměte přípravek z chladničky přibližně 30 minut před podáním.
Nejdříve připravte na aplikaci pacienta, potom si připravte podle následujících pokynů přípravek. Pokud není přípravek připraven správně podle pokynů pro přípravu, nesmí být podáván, protože v důsledku nesprávné rekonstituce přípravku může dojít k nedostatečnému klinickému účinku.
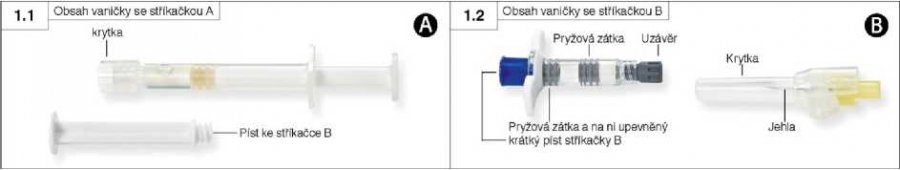
Krok 1: Otevřete obě vaničky (krycí fólii začněte strhávat z rohu, kde lze rozeznat malou bublinku) a vyprázdněte jejich obsah na čistou plochu (dvě vaničky obsahující stříkačku A (obrázek 1.1) a stříkačku B (obrázek 1.2)). Sáčky s vysoušedlem zlikvidujte.
Krok 2: Ze stříkačky B vytáhněte (ale nešroubujte) modře zbarvený krátký píst s připevněnou šedou zátkou a zlikvidujte jej (obrázek 2). Nepokoušejte se smísit přípravek, dokud je stříkačka opatřena oběma zátkami.

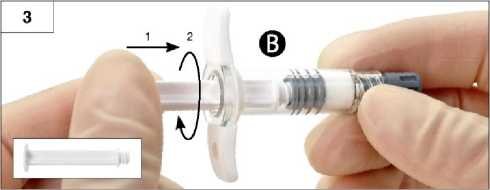
Krok 3: Jemně našroubujte bílý píst stříkačky B na šedou zátku, která zůstala ve stříkačce B (obrázek 3).
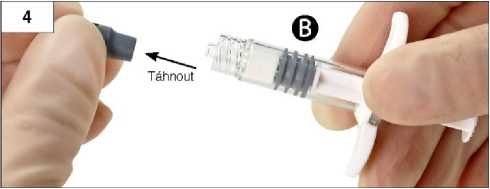
Krok 4: Vyjměte ze stříkačky B šedý pryžový uzávěr a stříkačku B odložte (obrázek 4).
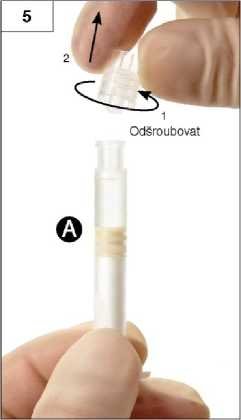
Krok 5: Uchopte stříkačku A do svislé polohy, abyste zabránili vytékání obsahu, a odšroubujte z ní průhlednou krytku (obrázek 5).
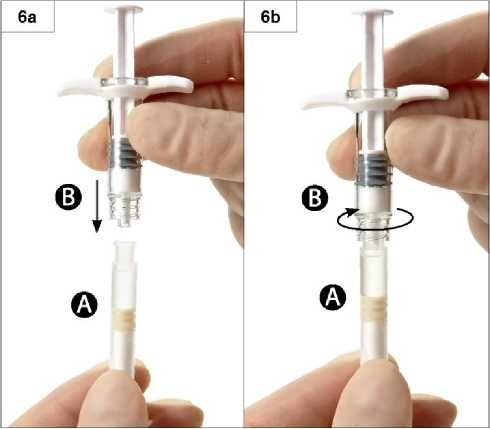
Krok 6: Spojte obě stříkačky tak, že jimi zatlačíte proti sobě a současně stříkačkou B pootočíte natolik, aby byly obě stříkačky bezpečně spojeny (obrázky 6a a 6b). Neutahujte více, než je nutné.
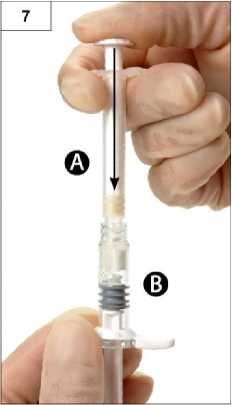
Krok 7: Spojené stříkačky obraťte opět do svislé polohy tak, aby byla stříkačka B dole. Současně vstříkněte kapalinu obsaženou ve stříkačce A do stříkačky B obsahující prášek (leuprorelin-acetát) (obrázek 7).
Krok 8: Jemným přetlačováním obsahu tam a zpět mezi stříkačkami ve vodorovné poloze přípravek důkladně promíchejte (celkem 60krát, což potrvá přibližně 60 sekund). Získáte homogenní, viskózní roztok (obrázek 8). Spojené stříkačky neohýbejte (mohlo by dojít k částečnému uvolnění závitu mezi stříkačkami a následnému úniku kapaliny).
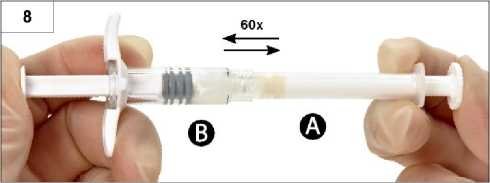
Po důkladném promíchání se bude barva vzniklého viskózního roztoku pohybovat na škále od bezbarvé přes bílou až po světle žlutou (může se vyskytnout bílé až světle žluté stínování).
Důležité: po promíchání přejděte okamžitě k dalšímu kroku, jelikož viskozita roztoku se s časem zvyšuje. Smíchaný přípravek chraňte před chladem.
Upozornění: promíchání přípravku musí být provedeno dle popisu, třepáním požadovaného stupně promíchání NEDOSÁHNETE.
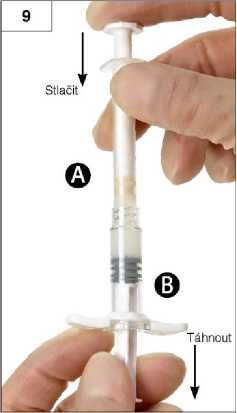
Krok 9: Uchopte stříkačky do svislé polohy, se stříkačkou B dole. Stříkačky mají být stále bezpečně spojené. Stlačením pístu stříkačky A a lehkým povytažením pístu stříkačky B natáhněte celý objem smíchaného přípravku do stříkačky B (krátká široká stříkačka) (obrázek 9).
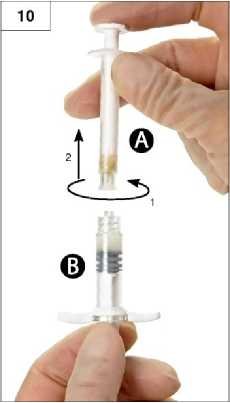
Krok 10: Nepřestávejte vyvíjet tlak na píst stříkačky A a současně tuto stříkačku odšroubujte
(obrázek 10). Zajistěte, aby nedošlo k úniku přípravku ze stříkačky. Nebylo by pak možné řádně upevnit injekční jehlu.
Upozornění: není na závadu, pokud ve směsi zůstane jedna velká nebo několik malých vzduchových bublin. Nesnažte se v tomto stádiu vzduchové bubliny ze stříkačky B odstranit, mohlo by to vést ke ztrátám přípravku!
Krok 11:
- Držte stříkačku B ve svislé poloze a zároveň přidržujte bílý píst, abyste zabránili ztrátě přípravku.
- Stržením zadního papírového štítku otevřete kontejner s bezpečnostní jehlou a jehlu vyjměte.
- Připevněte bezpečnostní jehlu ke stříkačce B tak, že budete držet stříkačku a jehlu na ni našroubujete jemným otáčením po směru hodinových ručiček asi o tři čtvrtě otáčky až do polohy, kdy jehla na stříkačku dosedne (obrázek 11).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Astellas Pharma s.r.o.
Sokolovská 100/94
186 00 Praha 8
Česká republika
8. REGISTRAČNÍ ČÍSLO(A)
44/078/05-C
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 23. 3. 2005
Datum posledního prodloužení: 13. 4. 2011
Další informace o léčivu ELIGARD
Jak
se ELIGARD
podává: subkutánní podání - prášek a rozpouštědlo pro injekční roztok
Výdej
léku: na lékařský předpis
Balení: Předplněná injekční stříkačka
Velikost
balení: 1+1 II
Držitel rozhodnutí o registraci daného léku v České republice:
Astellas Pharma s.r.o., Praha
E-mail: info@cz.astellas.com
Telefon: +420 236 080 300