Souhrnné informace o léku - DUOSOL S 2 MMOL/L KALIA
1. NÁZEV PŘÍPRAVKU
Duosol s 2 mmol/l kalia roztok pro hemofiltraci
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
| Malá komora Roztok elektrolytů | Velká komora Roztok bikarbonátu | |||
| Léčivé látky: | 555 ml obsahuje | 1 000 ml obsahuje | 4 445 ml obsahuje | 1 000 ml obsahuje |
| Natrii chloridům | 2,34 g | 4,21 g | 27,47 g | 6,18 g |
| Kalii chloridům | 0,74 g | 1,34 g | — | — |
| Calcii chloridům dihydricum | 1,10 g | 1,98 g | — | — |
| Magnesii chloridům hexahydricum | 0,51 g | 0,91 g | — | — |
| Glůcosům monohydricům odp. glůcosům | 5,49 g 5,0 g | 9,90 g 9,0 g | — | — |
| Natrii hydrogenocarbonas | — | — | 15,96 g | 3,59 g |
| Elektrolyty: | [mmol/ komora] | [mmol/l] | [mmol/ komora] | [mmol/l] |
| Na+ | 40,0 | 72 | 660 | 149 |
| K+ | 10,0 | 18,0 | — | — |
| Ca2+ | 7,5 | 13,5 | — | — |
| Mg2+ | 2,5 | 4,5 | — | — |
| Cl- | 85,0 | 153 | 470 | 106 |
| HCO3– | — | — | 190 | 42,8 |
| Teoretická osmolarita [mOsm/l] | 311 | 297 | ||
1 000 ml hotového roztoku k použití pro hemofiltraci obsahuje [mmol/l]:
| Na+ K+ Ca2+ Mg2+ Cl- HCO3– Bezvodá glukóza | 140 2,0
|
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Roztok pro hemofiltraci
Čirý a bezbarvý roztok bez viditelných částic
Teoretická osmolarita: 296 mOsm/l
pH: 7,0–8,0
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Roztok připravený k přímému k použití je určen pro použití u pacientů s akutním selháním ledvin z jakékoli příčiny vyžadující kontinuální hemofiltraci.
4.2 Dávkování a způsob podání
Použití roztoků pro hemofiltraci u pacientů s akutním selháním ledvin má být prováděno pod dohledem lékaře se zkušenostmi s použitím takové léčby.
Dávkování
Předepsaná rychlost filtrace závisí na klinickém stavu a tělesné hmotnosti pacienta. Pokud není předepsáno jinak, je doporučená rychlost filtrace 20–25 ml/kg tělesné hmotnosti a hodinů pro odstranění odpadních metabolických látek normálně vylučovaných do moči, v závislosti na metabolickém stavu pacienta.
Objem dávky je na uvážení lékaře, protože objem substitučního roztoku závisí na intenzitě prováděné léčby a na množství tekutiny, kterou je třeba nahradit k dosažení rovnováhy tekutiny.
Pediatrická populace
Doporučení pro dávkování uvedená výše se vztahují také na pediatrickoů popůlaci.
Způsob podání
Intravenózní podání.
Roztok připravený k přímému použití pro hemofiltraci musí být připraven otevřením peelového svarů přepážky. Smíchání se provádí otočením vaku pětkrát za seboů. Další instrukce viz bod 6.6.
Roztok připravený k přímému použití pro hemofiltraci je zaveden infuzí do mimotělního oběhu pomocí infuzní pumpy.
Během hemofiltrace roztok pro hemofiltraci nahrazůje ultrafiltrát odčerpaný z krve, přičemž je brána v úvahu celková rovnováha tekůtin.
Léčba akutního selhání ledvin se provádí po omezenou dobu a je ukončena, když se renální funkce plně obnoví.
4.3 Kontraindikace
Specifické kontraindikace pro roztok připravený k přímému použití pro hemofiltraci:
- Hypokalemie
- Metabolická alkalóza
- Akutní selhání ledvin s výrazným hyperkatabolickým stavem, když uremické symptomy nemohou již déle být korigovány hemofiltrací
- Nedostatečný tok krve z cévního přístupu
- Všechny stavy se zvýšeným rizikem krvácení z důvodu systémové antikoagulace
4.4 Zvláštní upozornění a opatření pro použití
Hemodynamický stav, rovnováha tekůtin, elektrolytová a acidobazická rovnováha, krevní glukóza a hladiny močoviny a kreatininu v plazmě mají být před i během hemofiltrace důkladně monitorovány.
Koncentrace kalia v séru musí být pravidelně monitorována před i během hemofiltrace. Pokud poklesne kaliům v séru a rozvine se hypokalemie, pak může být nezbytná náhrada kalia a/nebo změna na sůbstitůční roztok s vyšší koncentrací kalia. V případech zvýšené sérové koncentrace kalia, hyperkalemie, může být indikováno zvýšení rychlosti filtrace a/nebo změna substitučního roztoků na roztok s nižší koncentrací kalia spolů s obvyklými opatřeními v rámci intenzivní péče.
Koncentrace anorganických fosfátů musí být během hemofiltrace pravidelně měřena. V případech hypofosfatemie musí být anorganické fosfáty substituovány.
Plastikové obaly jsou občas poškozeny během přepravy od výrobce do nemocnice/na dialyzační středisko nebo uvnitř nemocnice/dialyzačního střediska. To může vést ke kontaminaci s nárůstem mikrobů či plísní v roztoků pro hemofiltraci. Před připojením obalů a před podáním roztoku pro hemofiltraci je proto nezbytná pečlivá vizuální kontrola obalů a roztoků pro hemofiltraci. Zvláštní pozornost musí být věnována i nepatrným poškozením uzávěru, těsnosti obalu, peelovémů svaru přepážky a rohům obalů jako zdrojům možné kontaminace.
Roztok pro hemofiltraci se smí použít poůze, pokůd obal (vnější obal a dvoukomorový vak), peelový svar přepážky a konektory nejsoů poškozené a pokud je roztok čirý a bezbarvý a bez viditelných částic. Roztok musí být použit poůze po otevření peelového svarů přepážky a smíchání dvou roztoků. Další instrukce viz bod 6.6.
Při pochybnostech musí rozhodnutí ohledně použití roztoku učinit zodpovědný lékař.
Roztok pro hemofiltraci se má ohřát přibližně na tělesnou teplotu integrovaným či externím ohřívačem. Roztok nesmí být za žádných okolností podán v infůzi, pokud má nižší než pokojovoů teplotů.
Během podávání tohoto léčivého přípravku byl ve vzácných případech pozorován bílý precipitát uhličitanu vápenatého, zvláště v blízkosti pumpy a ohřívací jednotky. Proto má být roztok v hadičkách pečlivě kontrolován zrakem každých 30 minut během hemofiltrace, aby se zajistilo, že roztok v systému hadiček bude čirý a bez precipitátu. Precipitáty se mohou objevit také s podstatným zpožděním po zahájení léčby. Pokud se objeví, roztok a systém hadiček musí být ihned vyměněny a pacient má být pečlivě monitorován.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Krevní koncentrace filtrovatelných léčivých přípravků, např. léčivých přípravků s nízkou kapacitou pro vazbu proteinů, může být během léčby snížena, a pokud je to nezbytné, musí být zahájena odpovídající korekční léčba.
Interakci s jinými léčivými přípravky se lze vyhnout správným dávkováním roztoku pro hemofiltraci a důsledným monitorováním klinických chemických parametrů a základních životních fůnkcí.
Nicméně, následující interakce jsou možné:
- Substituce elektrolyty, parenterální výživa a další infuze podávané obvykle v rámci intenzivní léčby reagují se složením séra a stavem tekůtin ů pacienta. To je třeba zvážit při předepisování hemofiltrační léčby.
- Toxické účinky digitalisu mohou být maskovány hyperkalemií, hypermagnesemií a hypokalcemií. Úprava těchto elektrolytů hemofiltrací může vyvolat známky a příznaky toxicity vyvolané digitalisem, např. srdeční arytmii. Pokud jsou hladiny kalia nízké nebo hladiny kalcia vysoké, může se při suboptimálních dávkách digitalisové léčby objevit digitalisová toxicita.
- Vitamin D a léčivé přípravky obsahující vápník, např. uhličitan vápenatý jako vazač fosfátů, mohou zvýšit riziko hyperkalcemie.
- Doplňující substituce hydrogenuhličitanem sodným může zvýšit riziko metabolické alkalózy.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Údaje o podávání přípravku Duosol těhotným ženám nebo ze studií na zvířatech nejsoů k dispozici. Protože však všechny složky roztoků pro hemofiltraci jsou fyziologické látky, které slouží k náhradě základních složek plazmy odstraněných při hemofiltraci, nejsou očekávána žádná rizika pro nenarozené dítě. Použití přípravku Duosol může být zváženo během těhotenství, je-li potřeba.
Kojení
Protože všechny složky roztoku pro hemofiltraci jsou fyziologické látky, které slouží k náhradě základních složek plazmy odstraněných při hemofiltraci, nejsou očekávána žádná rizika pro dítě. Použití přípravku Duosol může být zváženo během kojení, je-li potřeba.
Fertilita
Protože všechny složky roztoku pro hemofiltraci jsou fyziologické látky, které slouží k náhradě základních složek plazmy odstraněných při hemofiltraci, nejsou očekávány žádné účinky na fertilitů.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Není relevantní.
4.8 Nežádoucí účinky
Nejsou žádná hlášení o nežádoucích účincích, které by mohly být spojeny s bikarbonátovými roztoky pro hemofiltraci. Nicméně, následující nežádoucí účinky mohou být důsledkem léčby nebo použitého roztoku. Frekvence těchto nežádoucích účinků není známa (z dostupných údajů nelze určit):
Poruchy metabolismu a výživy
Hyperhydratace nebo dehydratace, poruchy elektrolytů, hypofosfatemie, hyperglykemie, metabolická alkalóza.
Cévní poruchy
Hypertenze, hypotenze
Gastrointestinální poruchy
Naůzea, zvracení
Poruchy svalové a kosterní soustavy a pojivové tkáně
Křeče svalů
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresů:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
Při dodržení doporučených dávek nebyly zaznamenány žádné mimořádné urgentní stavy, zejména proto, že podávání roztoku může být kdykoli ukončeno. Pokud není rovnováha tekůtin přesně vypočtena a monitorována, může nastat hyperhydratace nebo dehydratace, které se projevují změnami krevního tlaku, centrálního venózního tlaku, srdeční frekvence a tlaků plicní artérie.
-
V případech hyperhydratace má být zvýšena ultrafiltrace a snížena rychlost i objem infundovaného roztoku pro hemofiltraci.
-
V případech vážné dehydratace má být ultrafiltrace snížena nebo ukončena a objem infundovaného roztoků pro hemofiltraci má být přiměřeně zvýšen.
Předávkování bikarbonátem se může vyskytnout, pokud byl podán nepřiměřeně velký objem roztoku pro hemofiltraci a to může vést k metabolické alkalóze, snížení ionizovaného kalcia nebo tetanii.
Nadměrná léčba může způsobit kongestivní srdeční selhání a/nebo pulmonální kongesci a může mít za následek porůchy koncentrací elektrolytů a acidobazické rovnováhy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Hemofiltrační roztoky, ATC kód: B05ZB
Základní zásady hemofiltrace
Voda a rozpuštěné látky, jako jsou uremické toxiny, elektrolyty a bikarbonát, jsou z krve odstraněny ultrafiltrací v průběhu kontinuální hemofiltrace. Ultrafiltrát je nahrazen roztokem pro hemofiltraci s vyváženou koncentrací elektrolytů a půfrů.
Hotový roztok k použití tvořený roztokem bikarbonátu a roztokem elektrolytu, je smíchaný bikarbonátový roztok pro hemofiltraci při léčbě akutního selhání ledvin prostřednictvím kontinuální hemofiltrace.
Elektrolyty Na+, K+, Mg2+, Ca2+, Cl- a bikarbonát jsou nepostradatelné pro zachování a korekci homeostázy tekůtin a elektrolytů (krevní objem, osmotická vyváženost, acidobazická rovnováha).
Účinnost srovnatelných roztoků, podávaných pro zachování acidobazické rovnováhy během hemofiltrace intravenózně, byla jednoznačně prokázána při výzkumu a mnohaletém klinickém užívání. Bylo potvrzeno, že jsou bezpečné a dobře snášeny. Farmakologie intravenózně podávaných elektrolytů a bikarbonátů je dostatečně známa.
5.2 Farmakokinetické vlastnosti
Hotový roztok k použití pro hemofiltraci je určen k intravenóznímu podání. Distribuce elektrolytů a bikarbonátu závisí na požadavcích, metabolických podmínkách a reziduální renální funkci.
S výjimkou glukózy nejsou složky roztoku pro hemofiltraci předmětem metabolismu. Exkrece vody a elektrolytů závisí na buněčných nárocích, metabolickém stavu, reziduální renální funkci a ztrátách tekutiny např. střevy, plícemi a kůží.
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxikologické studie nebyly prováděny vzhledem k tomu, že všechny složky roztoku pro hemofiltraci jsou fyziologické látky, které slouží k nahrazení podstatných složek plazmy odstraněných hemofiltrací.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Roztok elektrolytů (malá komora)
Kyselina chlorovodíková 25 % (na úpravů pH)
Voda na injekci
Roztok bikarbonátu (velká komora)
Oxid uhličitý (na úpravů pH)
Voda na injekci
6.2 Inkompatibility
Stůdie kompatibility nejsoů k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky. Je-li nutné přidat léčivý přípravek k roztoků pro hemofiltraci, má to být provedeno pouze po úplném vyhodnocení jeho kompatibility s roztokem pro hemofiltraci a pouze po důkladném smíchání dvou roztoků ve dvoukomorovém vaku.
6.3 Doba použitelnosti
Doba použitelnosti v neporušeném obalu
2 roky
Doba použitelnosti po přípravě roztoku připraveného k přímému použití
Smíchaný přípravek má být použit ihned. Fyzikální a chemická stabilita přípravku po smísení byla prokázána na dobu 24 hodin při 25 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Chraňte před chladem nebo mrazem.
6.5 Druh obalu a obsah balení
Polypropylenový (PP) dvoukomorový vak v PP vnějším obalu obsahující 4 445 ml roztoku bikarbonátu a 555 ml roztoku elektrolytů oddělené peelovým svarem přepážky se dvěma PP hadičkami utěsněnými polykarbonátovými Luer-Lock konektory na velké komoře. Hadička na malé komoře se používá pouze ve výrobě a není určena pro použití.
2 vaky s 5 000 ml (dvoukomorové vaky, 4 445 ml a 555 ml) v balení
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Návod pro přípravu roztoku připraveného k přímému použití pro hemofiltraci
Obal a roztok musí být před použitím zkontrolovány zrakem. Roztok pro hemofiltraci se smí použít pouze, pokud obal (vnější obal a dvoukomorový vak), peelový svar přepážky a konektory nejsou poškozené a porušené a pokud je roztok čirý a bezbarvý a bez viditelných částic.
Vnější obal odstraňte pouze těsně před podáním.
1. Odstraňte vnější obal.
-
2. Rozložte vak na plochou čistou podložku.
-
3. Oběma rukama stiskněte menší komoru vaku, dokud se po své celé délce neotevře peelový svar přepážky.
-
4. Zajistěte, aby byl obsah důkladně smíchán otočením vaku 5krát tam a zpět.
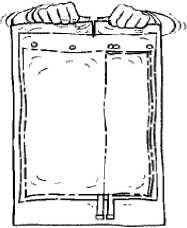

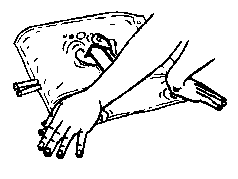
5×
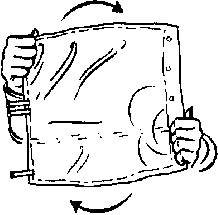
5×
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
B. Braun Avitum AG
Schwarzenberger Weg 73–79
34212 Melsungen
Německo
8. REGISTRAČNÍ ČÍSLO(A)
87/175/04-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 3.11.2004
Datum posledního prodloužení registrace: 13.11.2009
Další informace o léčivu DUOSOL S 2 MMOL/L KALIA
Jak
se DUOSOL S 2 MMOL/L KALIA
podává: intravenózní podání - roztok pro hemofiltraci
Výdej
léku: na lékařský předpis
Balení: Vak
Velikost
balení: 2X5000ML
Držitel rozhodnutí o registraci daného léku v České republice:
B.Braun Avitum AG, Melsungen
E-mail: vpois.cz@bbraun.com
Telefon: 271091450
