Souhrnné informace o léku - BELKYRA 10 MG/ML INJEKČNÍ ROZTOK
1. NÁZEV PŘÍPRAVKU
Belkyra 10 mg/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml injekčního roztoku obsahuje acidum deoxycholicum 10 mg.
Jedna injekční lahvička obsahuje acidum deoxycholicum 20 mg ve 2 ml roztoku.
Pomocná látka/pomocné látky se známým účinkem
Jeden ml obsahuje 184 |jmol (nebo 4,23 mg) sodíku z chloridu sodného, hydroxidu sodného a hydrogenfosforečnanu sodného.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Čirý, bezbarvý roztok bez viditelných částic.
pH přípravku je upraveno na 8,3 pomocí kyseliny chlorovodíkové nebo hydroxidu sodného a jeho osmotický tlak je kompatibilní s biologickými tkáněmi a tekutinami s osmolalitou 300 mosm/kg.
| 4. | KLINICKÉ ÚDAJE |
| 4.1 | Terapeutické indikace |
| – | Přípravek Belkyra je indikován k léčebnému výkonu v oblasti středně závažného |
| až závažného vyklenutí nebo plnosti spojené s přítomností submentálního tuku u dospělých, kdy má submentální tuk psychologický dopad na pacienta | |
| 4.2 | Dávkování a způsob podání |
Dávkování
Celkový injekčně aplikovaný objem a počet léčebných výkonů má být stanoven individuálně podle rozložení submentálního tuku a léčebných cílů každého pacienta.
Injekčně se aplikuje 0,2 ml (2 mg) na jedno místo vpichu s odstupem 1 cm. Při jednom léčebném úkonu nemá být překračována maximální dávka 10 ml (100 mg odpovídající 50 injekcím).
Lze provést maximálně 6 léčebných úkonů. U většiny pacientů dochází ke zlepšení po 2 až 4 léčebných výkonech.
Časový interval mezi léčebnými výkony má být alespoň 4 týdny.
V zájmu většího pohodlí pacienta lze při aplikaci přípravku na základě rozhodnutí zdravotnického pracovníka podat perorální analgetika nebo NSAID, lokální a/nebo injekční lokální anestezii (např. lidokain) a oblast aplikace přípravku chladit pomocí sáčků s ledovým gelem.
Zvláštní skupiny pacientů
Porucha funkce ledvin
Není zapotřebí provádět žádnou úpravu dávky (viz bod 5.2).
Porucha _ funkce , jater
Není zapotřebí provádět žádnou úpravu dávky (viz bod 5.2).
Starší pacienti (65 a více let)
Není zapotřebí provádět žádnou úpravu dávky. U starších pacientů je zapotřebí postupovat s opatrností. (viz bod 4.4).
Pediatrická populace
Použití přípravku Belkyra u dětí nebo dospívajících není relevantní.
Způsob podání
Přípravek je určení výhradně k subkutánnímu podání.
Přípravek Belkyra mají podávat pouze zdravotničtí pracovníci s odpovídající kvalifikací, vědomostmi a znalostmi submentální anatomie. Bezpečné a účinné použití přípravku Belkyra závisí na výběru vhodných pacientů, což zahrnuje znalost dřívějších zákroků v anamnéze pacienta a jejich potenciálu umožňujícímu změny této anatomické oblasti. Pečlivě zvažte použití přípravku Belkyra u pacientů s nadměrně volnou kůží, výraznými pásmy platysmy nebo jinými stavy, u kterých může redukce submentálního tuku vést k nežádoucímu výsledku.
Přípravek Belkyra má být používán pouze k jednomu léčebnému výkonu spočívajícímu v podání injekce/injekcí pacientovi; nepoužitý přípravek je třeba řádně zlikvidovat.
Belkyra se dodává v jednorázových injekčních lahvičkách připravených k použití. Před použitím injekční lahvičku několikrát opatrně obraťte dnem vzhůru. Neřeďte.
Při aplikaci injekce s přípravkem Belkyra umístěte jehlu kolmo k pokožce.
Umístění jehly vůči čelisti je velmi důležité, protože snižuje riziko poranění okrajového čelistního nervu, motorické větve obličejového nervu. Poranění nervu se projevuje nesymetrickým úsměvem způsobeným parézou depresoru rtu.
Dodržením následujících pokynů se předejde poranění části mandibulárního nervu:
- neaplikujte injekci nad dolní okraj mandibuly.
- neaplikujte injekci do oblasti definované 1–1,5cm linií pod dolním okrajem mandibuly (od úhlu mandibuly k bradě).
- aplikujte přípravek Belkyra injekčně v cílové oblasti submentálního tuku (viz obrázky 1 a 3).
Obrázek 1. Vyhněte se oblasti okrajového mandibulárního nervu

Neaplikujte injekci do platysmatu. Před každým léčebným výkonem prohmatejte submentální oblast, ověřte, že je množství submentálního tuku dostatečné, a zjistěte subkutánní tuk mezi dermis a platysmatem (preplatysmální tuk) v cílové oblasti léčebného výkonu (obrázek 2).
Obrázek 2. Předozadní pohled na oblast platysmatu
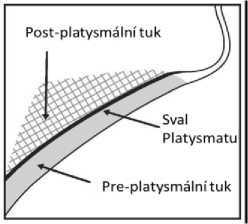
Ohraničte oblast plánovaného léčebného výkonu chirurgickým fixem a aplikací 1cm2 injekční mřížky označte místa aplikace injekce (obrázky 2 a 3).

Nevpichujte přípravek Belkyra mimo definované parametry.
Injekční roztok je třeba před aplikací vizuálně zkontrolovat. Použít se mohou pouze bezbarvé roztoky bez viditelných částic.
4.3 Kontraindikace
-
– Hypersenzitivita na kyselinu deoxycholovou nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1
-
– Přítomnost infekce v navrhovaných místech aplikace injekce
4.4 Zvláštní upozornění a opatření pro použití
Pouze subkutánní podání.
Injekce do citlivých oblastí nebo jejich blízkosti
Nevpichujte do vzdálenosti 1 až 1,5 cm od citlivých anatomických struktur.
Přípravek Belkyra nemá být aplikován do okraje čelistní větve lícního nervu nebo v její blízkosti, aby nenastala možnost motorické neurapraxie, která se projevuje jako nesymetrický úsměv nebo ochablost obličejových svalů. V klinických studiích bylo poranění lícního nervu dočasné a ve všech případech došlo k úpravě.
Je třeba dbát na to, aby nedošlo k nežádoucímu intradermálnímu nebo intramuskulárnímu vpichu. Přípravek Belkyra má být injekčně aplikován do středu preplatysmální subkutánní tukové tkáně v submentální oblasti. V průběhu aplikace injekce nevytahujte jehlu ze subkutánního tuku, protože by se tak mohlo zvýšit riziko intradermální expozice a potenciální ulcerace pokožky.
Zabraňte aplikaci injekce do slinných žláz, štítné žlázy, lymfatických uzlin a svalů.
Bezpečné a účinné použití přípravku Belkyra mimo oblast submentálního tuku nebylo stanoveno.
Stávající onemocnění/léčebné výkony v léčené oblasti nebo , její blízkosti
U pacientů je před použitím přípravku Belkyra třeba provést screening potenciálních příčin submentálního vyklenutí/plnosti (např. thyromegalie a cervikální lymfadenopatie).
Při podávání přípravku Belkyra je třeba postupovat s opatrností v přítomnosti zánětu nebo indurace v navrhovaných místech vpichu injekce nebo u pacientů s projevy dysfagie.
Je třeba postupovat s opatrností, pokud je přípravek Belkyra podáván pacientům, kteří dříve podstoupili chirurgický nebo estetický léčebný výkon v submentální oblasti. Změny anatomie/orientačních bodů nebo přítomnost zjizvené tkáně mohou mít vliv na schopnost bezpečně aplikovat přípravek Belkyra nebo dosáhnout žádoucích výsledků.
Starší pacienti
Klinické studie s přípravkem Belkyra nezahrnovaly dostatečný počet pacientů ve věku 65 a více let ke stanovení toho, zda reagují odlišně než mladší pacienti; proto je u těchto pacientů zapotřebí postupovat s opatrností.
Nízkosodíková dieta
Tento přípravek obsahuje 184 ^mol (nebo 4,23 mg) sodíku na 1 ml. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné klinické studie lékových interakcí s přípravkem Belkyra.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Byly provedeny reprodukční studie na potkanech a králících s expozicí až 1,8krát (potkan) a 12krát (králík) vyšší, než je maximální doporučovaná dávka u lidí. Ačkoli tyto studie neprokázaly přímé ani nepřímé škodlivé účinky, co se týče reprodukční toxicity, v embryonálně-fetální studii toxicity (viz bod 5.3) byly zjištěny neprůkazné závěry o chybějícím středním plicním laloku.
Nebyly provedeny žádné adekvátní a dobře kontrolované studie u těhotných žen. V rámci bezpečnostních opatření se nemá přípravek Belkyra během těhotenství používat.
Kojení
K dispozici nejsou žádné informace o přítomnosti kyseliny deoxycholové v lidském mléce, účincích tohoto přípravku na kojené dítě ani účincích tohoto léku na produkci mléka. Protože studie s kojícími matkami nebyly provedeny, je třeba postupovat při podávání přípravku Belkyra kojícím ženám s opatrností.
Fertilita
Nejsou k dispozici žádné klinické údaje o fertilitě.
Přípravek Belkyra neovlivňoval obecný reprodukční výkon ani fertilitu u samců ani samic potkanů v dávkách až 50 mg/kg, což odpovídá přibližně 5násobku, respektive 3násobku maximální doporučené dávky u lidí (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie schopnosti řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Údaje popsané v tabulce níže odrážejí nežádoucí účinky hlášené u pacientů léčených přípravkem Belkyra (n=1118), kteří byli hodnoceni v klinických studiích s přípravem Belkyra v redukci submentálního tuku.
Následující nežádoucí účinky byly hodnoceny v klinických studiích s těmito četnostmi:
| (> 1/10) (> 1/100 až < 1/10) (> 1/1 000 až < 1/100) (> 1/10 000 až < 1/1 000) (< 1/10 000) (z dostupných údajů nelze určit). |
| Třída orgánových systémů | Frekvence | Nežádoucí účinky |
| Poruchy nervového systému | Časté | Bolest hlavy |
| Méně časté | Dysgeuzie | |
| Respirační, hrudní a mediastinální poruchy | Méně časté | Dysfonie |
| Gastrointestinální poruchy | Časté | Dysfagie, nauzea |
| Poruchy kůže a podkožní tkáně | Časté | Napnutí kůže |
| Celkové poruchy a reakce v místě aplikace | Velmi časté | Místo vpichu: bolest, edém, zduření, necitlivost, uzliny, |
| hematom, parestezie, indurace, erytém, pruritus. | ||
| Časté | Místo vpichu: krvácení, diskomfort, pocit horka, změna barvy. | |
| Méně časté | Místo vpichu: alopecie, urtikarie, vřed, hypersenzitivita. | |
| Poranění, otravy a procedurální komplikace | Časté | Poranění nervu v místě vpichu |
Hlášení podezření na nežádoucí reakce
Hlášení podezření na nežádoucí reakce po registraci léčivého přípravku je důležité. Umožňuje pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
webové stránky:
4.9 Předávkování
Nebylo hlášeno předávkování s přípravkem Belkyra u lidí.
Lze očekávat, že zvýšení objemu injekcí nebo zmenšení odstupu mezi injekcemi přípravku Belkyra může zvýšit riziko lokálních nežádoucích účinků. Reakce neléčené oblasti nebo systémové nežádoucí účinky byly v klinických studiích dávky až 200 mg vzácné.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakologické vlastnosti
Farmakoterapeutická skupina: jiná dermatologika
Kód ATC: D11AX24
Mechanismus účinku
Kyselina deoxycholová je cytolytickým lékem, který při injekční aplikaci do lokalizovaného subkutánního tuku fyzicky narušuje buněčnou membránu adipocytů. Zničení adipocytů vyvolá tkáňovou reakci, při které jsou do dané oblasti přitahovány makrofágy eliminující buněčný odpad a tuky, které jsou poté zlikvidovány přirozenými procesy. Poté dochází ke vzniku fibroblastů a pozorovanému ztluštění fibrózního septa, což naznačuje nárůst celkového kolagenu (tj. neokolagenózu).
Klinická účinnost a bezpečnost
Byly provedeny čtyři randomizované, multicentrické, dvojitě zaslepené, placebem kontrolované studie fáze 3 (2 identické studie prováděné v Evropské unii [EU] a 2 identické studie prováděné v Severní Americe) hodnotící přípravek Belkyra v léčebném výkonu v oblasti vyklenutí nebo plnosti spojené se submentálním tukem (SMF) a hodnocení spojeného psychologického dopadu. Ve všech studiích byly primární cílové parametry měřeny 12 týdnů po posledním léčebném výkonu. Všechny studie fáze 3 splnily své primární cílové parametry účinnosti a ukázaly zlepšení v psychologickém dopadu oproti placebu.
Do těchto studií byli přijímáni dospělí pacienti (věk 19 až 65 let) se středním až závažným zaoblením nebo plností spojenými se submentálním tukem (tj. stupeň 2 nebo 3 na 5bodové škále, kde 0 = nejsou přítomny, 4 = extrémní) jak podle úsudku lékaře, tak hodnocení samotných pacientů. Pacientům byly provedeny až 4 léčebné výkony ve studiích prováděných v EU a až 6 výkonů ve studiích prováděných v Severní Americe s přípravkem Belkyra (n=757 pro všechny 4 studie) nebo s placebem (n=746) v 28denních intervalech. Léčebný výkon byl ukončen po dosažení požadovaných výsledků. Objem injekcí činil 0,2 ml na jedno místo vpichu s rozestupem 1 cm do submentální tukové tkáně, což lze také vyjádřit jednotkou dávky na jednotku plochy 2 mg/cm2. Při každém výkonu bylo povoleno maximálně 100 mg (10 ml) na celkovou léčenou oblast.
Průměrný věk pacientů ve studiích prováděných v EU byl 46 let a průměrný BMI byl 26. Většinu pacientů představovaly ženy (75 %) a většina pacientů byla bělošského etnika (94 %). Ve vstupním stavu mělo podle posouzení lékaře 68 % pacientů středně závažný a 32 % velmi závažný výskyt submentálního tuku. U studií provedených v Severní Americe byl průměrný věk 49 let a průměrný BMI činil 29 kg/m2. Většinu pacientů představovaly ženy (85 %) a většina pacientů byla bělošského etnika (87 %). Na začátku léčebného výkonu mělo podle posouzení lékaře 51 % pacientů středně závažný a 49 % velmi závažný výskyt submentálního tuku.
Koprimárními hodnoceními účinnosti ve studiích prováděných v EU bylo hodnocení submentálního tuku uváděné lékařem (CR-SMFRS) a pacientovo hodnocení vlastní spokojenosti (SSRS). Hodnocení submentálního tuku uváděné samotným pacientem (PR-SMFRS) bylo rovněž posuzováno. Psychologický dopad submentálního tuku byl hodnocen pomocí několika ukazatelů včetně Derrifordovy škály vzhledu 24 (DAS-24), Body Image Quality of Life Inventory (BIQLI) a 6položkového dotazníku škály dopadu submentálního tuku podle hlášení pacienta (PR-SMFIS) (hodnotícího štěstí, obtěžování, nesmělost, rozpaky, dojem staršího vzhledu nebo nadváhy). Statisticky významná zlepšení podle hodnocení lékaře i pacienta, spokojenost pacientů a snížení psychologického dopadu submentálního tuku byly častěji pozorovány ve skupině s přípravkem Belkyra než ve skupině s placebem (tabulka 1). Snížení objemu submentálního tuku bylo potvrzeno měřením pomocí kaliperu.
Ve studiích provedených v Severní Americe byla koprimární hodnocení účinnosti založena na zlepšení submentálního zaoblení nebo plnosti alespoň o 2 stupně nebo alespoň o 1 stupeň v lékařem udávaném (CR-SMFRS) a pacientem udávaném (PR-SMFRS) hodnocení submentálního tuku 12 týdnů po posledním léčebném výkonu. Psychologický dopad SMF byl hodnocen pomocí stejného 6položkového dotazníku jako ve studiích v EU. Kromě toho byly změny v submentálním tuku hodnoceny na podmnožině pacientů (n=449, kombinované studie) pomocí magnetické rezonance (MRI). Snížení objemu submentálního tuku bylo potvrzeno jak MRI snímkováním, tak kaliperem.
Tabulka 1 níže zobrazuje zlepšení jednostupňové lékařské reakce (CR-SMFRS), reakce spokojenosti pacienta (SSRS) a psychologického dopadu (PR-SMFIS), které se vztahují na všechny čtyři studie fáze 3. Obrázek 4 uvádí podíly odpovědi založené na hodnocení submentálního tuku lékařem při každé návštěvě v rámci studie.
Tabulka 1: Hodnocení submentálního tuku lékařem a pacientem, spokojenost a psychologický dopad 12 týdnů po posledním léčebném výkonu
| Studie prováděné v EU a | Studie prováděné v Severní Americe b | |||
| Cílový parametr | BELKYRA (n=243) | Placebo (n=238) | BELKYRA (n=514) | Placebo (n=508) |
| Jednostupňová reakce lékaře (CR-SMFRS) c | 63,8 % | 28,6 % | 78,5 % | 35,3 % |
| Jednostupňová reakce pacienta (PR-SMFRS) c | 63,1 % | 34,3 % | 80,3 % | 38,1 % |
| Reakce spokojenosti pacienta (SSRS) d | 65,4 % | 29 % | 69,1 % | 30,5 % |
| Průměrné procentuální zlepšení psychologického dopadu (PR-SMFIS) ve srovnání se vstupním stavem e | 44,6 % | 18,0 % | 48,6 % | 17,3 % |
a Povoleny až 4 léčebné výkony b Povoleno až 6 léčebných výkonů
c Zlepšení redukce v lékařem hlášeném submentálním tuku (CR-SMFRS) alespoň o jeden stupeň 12 týdnů po posledním léčebném výkonu
d Pacientovo hodnocení „velice spokojený“, „spokojený“ nebo „mírně spokojený“ na SSRS 12 týdnů po posledním léčebném výkonu
e Průměr procentuálního zlepšení od vstupního stavu vypočítaný jako průměrná změna PR-SMFIS od vstupního stavu vydělená průměrem ve vstupním stavu
| Obrázek 4: | Hodnocení submentálního tuku lékařem (CR-SMFRS) Podíly jednostupňových respondérů na každé studijní návštěvě; souhrnné údaje z klinických studií v EU (levý panel) a Severní Americe (pravý panel) |
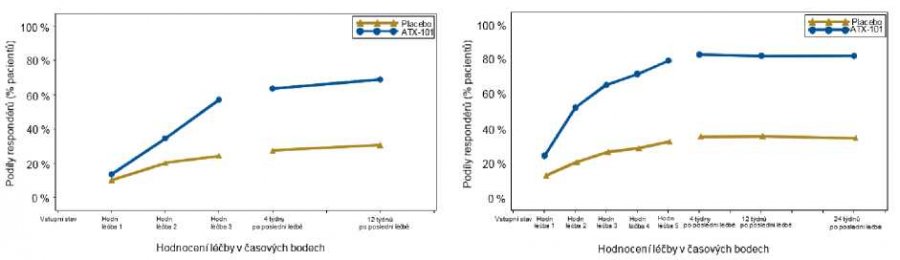
p < 0,001 pro všechny časové body, BELKYRA vs. placebo
Navzdory tomu, že u většiny pacientů došlo k redukci objemů submentálního tuku, 90,0 % pacientů ve studiích v EU a 92 % pacientů ve studiích v USA/Kanadě nevykázalo žádnou změnu (68,9 % a 70,5 %) ani zlepšení (21,6 % a 22,9 %) ve skóre pružnosti kůže 12 týdnů po posledním ošetření ve srovnání se začátkem léčby.
Dlouhodobá bezpečnost a udržení léčebného účinku byly hodnoceny po ošetření přípravkem Belkyra. Podmnožina počátečních respondérů léčených přípravkem Belkyra pokračovala v těchto následných studiích, kde bylo udržení léčebného účinku prokázáno až na 5 let.
Pediatrická populace
Použití přípravku Belkyra u osob mladších 18 let se nedoporučuje.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předkládat výsledky studií přípravku Belkyra pro všechny podskupiny pediatrické populace v léčebném výkonu v oblasti středně závažného až závažného vyklenutí nebo plnosti spojené s přítomností submentálního tuku u dospělých, kdy má submentální tuk psychologický dopad na pacienta (viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Endogenní plazmatické hladiny kyseliny deoxycholové jsou velmi proměnlivé u každého jednotlivce i mezi nimi; většina této přirozené sekundární žlučové kyseliny je oddělena v enteropatickém oběhovém systému. Farmakokinetika exogenní kyseliny deoxycholové podávané léčebným výkonem přípravku Belkyra byla srovnána s tímto endogenním prostředím.
Absorpce
Kyselina deoxycholová obsažená v přípravku Belkyra se po subkutánní injekci rychle absorbuje. Po podávání maximální doporučené dávky pro jedno ošetření přípravkem Belkyra (100 mg) byly pozorovány maximální koncentrace v plazmě (mean Cmax) s mediánem tmax 6 minut po injekci. Průměrná hodnota Cmax byla 1 036 ng/ml a byla 2,3krát vyšší než průměrné hodnoty Cmax pozorované během 24hodinového vstupního endogenního období v nepřítomnosti přípravku Belkyra. Při maximální doporučené dávce pro jedno ošetření (100 mg) byla expozice kyselině deoxycholové (AUC0–24) méně než 2krát vyšší než endogenní expozice. Plazmatická hodnota
AUC0–24 rostla způsobem odpovídajícím dávce až 100 mg. Plazmatické hladiny kyseliny deoxycholové po léčebném výkonu se navrátily k endogennímu rozsahu během 24 hodin. Při navrhovaném intervalu léčebných výkonů se neočekává žádná akumulace.
Distribuce
Distribuční objem byl odhadnut na 193 l a nezávisí na dávce až do 100 mg. Deoxycholová kyselina se extenzivně váže na proteiny v plazmě (98 %).
Eliminace
Endogenní kyselina deoxycholová je produktem metabolismu cholesterolu a v nezměněném stavu se vylučuje stolicí. Kyselina deoxycholová obsažená v přípravku Belkyra se spojuje se žlučovými kyselinami a je vylučována společně s endogenní kyselinou deoxycholovou. Kyselina deoxycholová je eliminována hepatickými transportními proteiny z krve do žluče, aniž by jakkoli přispívala k metabolismu.
Kyselina deoxycholová není in vitro inhibitorem enzymů CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 ani 3A4. Kyselina deoxycholová na klinické úrovni neindukovala CYP1A, 2B6 ani 3A.
Kyselina deoxycholová není in vitro inhibitorem transportérů BSEP, MRP2, MRP4, MDR1, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, OATP2B1 ani ASBT. Kyselina deoxycholová inhibovala NTCP a IC50 2,14 in vitro.
Porucha funkce ledvin
Přípravek Belkyra nebyl studován u pacientů s poruchou funkce ledvin. Žlučové kyseliny včetně kyseliny deoxycholové jsou do moči vylučovány v zanedbatelných množstvích; není pravděpodobné, že by porucha funkce ledvin ovlivňovalo farmakokinetiku kyseliny deoxycholové.
Porucha funkce jater
Přípravek Belkyra nebyl studován u pacientů s poruchou funkce jater. S ohledem na přerušovanou frekvenci dávkování, malou dávku, která představuje přibližně 3 % celkového množství žlučových kyselin, jakož i na vysoce variabilní hladiny endogenní kyseliny deoxycholové je nepravděpodobné, že by byla farmakokinetika kyseliny deoxycholové po aplikaci injekce přípravku Belkyra ovlivněna poruchou funkce jater.
Starší pacienti
Není zapotřebí provádět žádnou úpravu dávky. U starších pacientů je zapotřebí postupovat s opatrností (viz bod 4.4).
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity, reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
Kancerogenita
Ve studiích toxicity po opakovaném dávkování trvajícím 6 měsíců u potkanů a 9 měsíců u psů nebyly zjištěny žádné indikace lokálních nebo systemických pre-neoplastických odpovědí na subkutánní podání přípravku Belkyra. V těchto studiích byla maximální zamýšlená klinická dávka překročena 2,5krát u potkanů až 12,5krát u psů (na základě mg/místo vpichu) a 2krát u potkanů až 4krát u psů (na základě kvantifikované systemické expozice). Dále navzdory maximálnímu zamýšlenému klinickému režimu měsíčních injekcí po až 6 léčebných výkonů byly injekce přípravku Belkyra aplikovány dvakrát měsíčně až po celkem 13 dávek u potkanů a celkem 20 dávek u psů. S přípravkem Belkyra nebyly provedeny žádné studie karcinogenity.
Genotoxicita
Přípravek Belkyra byl negativní v in vitro (analýza mikrobiální reverzní mutace a analýza chromozomální aberace) a in vivo (analýza mikrojadérka) genetických toxikologických analýzách.
Vývojová toxicita
V embryonálně-fetální studii toxicity byly zjištěny neprůkazné závěry o chybějícím středním plicním laloku. Tento závěr byl signifikantně vyšší u skupiny s dávkou 30 mg/kg, ale byl patrný také při nejnižší koncentraci 10 mg/kg. Tato dávka byla spojena s mateřskou lokální toxicitou. Klinický význam tohoto závěru není zřejmý.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Voda na injekci
Chlorid sodný
Hydroxid sodný (na úpravu pH)
Hydrogen fosforečnan sodný
Kyselina chlorovodíková (na úpravu pH)
6.2 Inkompatibilita
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
30 měsíců
Tento přípravek má být použit bezprostředně po proděravění zátky injekční lahvičky.
Pokud není použit okamžitě, jsou podmínky a doba skladování při používání na odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání po prvním otevření léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
Injekční roztok v injekční lahvičce (sklo třídy I) opatřené zátkou (chlorbutylová pryž) a uzávěrem (hliník) s odklápěcím víčkem (polypropylen).
Jedna krabička obsahuje 4 injekční lahvičky. Jedna injekční lahvička obsahuje 2 ml injekčního roztoku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro manipulaci s ním
Každá injekční lahvička je určena pouze k jednorázovému použití. Po použití je třeba veškerý nepoužitý přípravek zlikvidovat.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals International Ltd
Clonshaugh Industrial Estate
Dublin 17 – Coolock
Irsko
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
87/406/16-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19.10.2016
Další informace o léčivu BELKYRA 10 MG/ML INJEKČNÍ ROZTOK
Jak
se BELKYRA 10 MG/ML INJEKČNÍ ROZTOK
podává: subkutánní podání - injekční roztok
Výdej
léku: na lékařský předpis
Balení: Injekční lahvička
Velikost
balení: 4X2ML
Držitel rozhodnutí o registraci daného léku v České republice:
Allergan Pharmaceuticals International Limited, Dublin
Telefon: 800 18 88 18