Souhrnné informace o léku - AZELASTIN-POS 1 MG/ML NOSNÍ SPREJ, ROZTOK
1. NÁZEV PŘÍPRAVKU
AZELASTIN-POS 1 mg/ml nosní sprej, roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Roztok obsahující azelastini hydrochloridum 1 mg/ml Podaná dávka na jedno stlačení (0,14 ml) obsahuje azelastini hydrochloridum 0,14 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Nosní sprej, roztok. Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Symptomatická léčba sezónní alergické rinitidy. AZELASTIN-POS nosní sprej je indikován u dospělých a dětí od 6 let věku.
4.2 Dávkování a způsob podání
Dávkování
Jedno vstříknutí přípravku AZELASTIN-POS nosní sprej do každé nosní dírky dvakrát denně (ráno a večer). To odpovídá denní dávce azelastini hydrochloridum 0,56 mg.
Pediatrická populace
AZELASTIN-POS nosní sprej je vhodný u dospělých a dětí od 6 let věku. Přípravek AZELASTIN-POS nosní sprej se nemá používat u dětí do 6 let věku kvůli chybějícím údajům o bezpečnosti a účinnosti.
Způsob podání
Nosní podání.
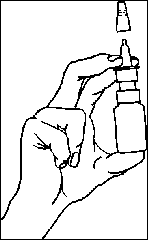
Před použitím odstraňte uzávěr.
Stiskněte dávkovací mechanismus, dokud nedojde k vytlačení roztoku (obvykle jednou nebo dvakrát). Systém je nyní připraven k použití pro jakékoliv další použití.
Zasuňte nosní adaptér (trysku) do nosní dírky a jednou stiskněte, současně lehce dýchejte. Aplikujte jedno vstříknutí do každé nosní dírky. Sprej aplikujte s hlavou ve svislé poloze, během podání nenaklánějte hlavu daleko dozadu. Z hygienických důvodů otřete nosní adaptér po každé aplikaci a uzavřete jej ochrannou krytkou.
AZELASTIN-POS nosní sprej má používat pouze jedna a tatáž osoba.
Délka trvání léčby
AZELASTIN-POS nosní sprej lze používat, odkud obtíže nezmizí, ale nemá se používat bez přerušení déle než 6 měsíců.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku azelastini hydrochloridum nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Pediatrická populace
Přípravek AZELASTIN-POS se nemá používat u dětí do 6 let věku.
4.4 Zvláštní upozornění a opatření pro použití
AZELASTIN-POS nosní sprej není vhodný pro léčbu běžného nachlazení nebo chřipky.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Do dnešní doby nejsou známy žádné interakce u lokálního použití azelastini hydrochloridum.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Údaje o podávání azelastinu těhotným ženám jsou omezené nebo nejsou k dispozici. Použití přípravku AZELASTIN-POS nosní sprej se nedoporučuje během prvního trimestru těhotenství, přestože nejsou žádné důkazy o teratogenním účinku na zvířatech i při dávkách vyšších než používaných k léčebným účelům.
Kojení
AZELASTIN-POS nosní sprej se během kojení nemá podávat kvůli nedostatečným údajům.
4.7 Účinky na schopnost řídit a obsluhovat stroje
V některých případech po použití přípravku AZELASTIN-POS nosní sprej mohou pacienti trpět únavou, vyčerpaností, vyčerpáním, závratěmi nebo slabostí, což jsou obtíže, které může způsobit i onemocnění samotné. V těchto případech může být narušena schopnost řídit a obsluhovat stroje. Zvláštní pozornost je nutné věnovat tomu, že alkohol nebo jiné léčivo může tyto účinky zesilovat.
4.8 Nežádoucí účinky
Nežádoucí účinky jsou uvedené níže podle třídy orgánových systémů a frekvence. Frekvence jsou definovány takto:
Velmi časté (> 1/10);
Časté (> 1/100 až < 1/10);
Méně časté (> 1/1 000 až < 1/100);
Vzácné (> 1/10 000 až < 1/1 000);
Velmi vzácné (<1/10000);
Není známo (z dostupných údajů nelze určit).
Poruchy imunitního systému
Velmi vzácné: Hypersenzitivní reakce
Poruchy nervového systému
Časté: Dysgeuzie (nepříjemná chuť) se může objevit po podání (často kvůli nesprávné metodě podání, konkrétně naklonění hlavy příliš daleko dozadu během podání), které může vést ve vzácných případech k nauzee.
Velmi vzácné: Závratě
Respirační, hrudní a mediastinální poruchy
Méně časté: Nosní diskomfort zanícené nosní tkáně (bodání, svědění), kýchání, epistaxe
Gastrointestinální poruchy
Vzácné: Nauzea
Celkové poruchy a reakce v místě aplikace
Velmi vzácné: Únava (vyčerpanost, vyčerpání), závratě nebo slabost
Poruchy kůže a podkožní tkáně
Velmi vzácné: Vyrážka, pruritus, urtikárie
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
S podáváním toxických dávek azelastini hydrochloridum u člověka nejsou k dispozici žádné zkušenosti.
Příznaky:
V případě předávkování nebo intoxikace se na základě výsledků při pokusech na zvířatech očekávají poruchy centrálního nervového systému.
Léčba:
Léčba těchto poruch musí být symptomatická. Není známo žádné antidotum.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Dekongesční a jiná nosní léčiva k lokální aplikaci; antialergika kromě kortikosteroidů; ATC kód: R01AC03
Azelastin je klasifikován jako silné, dlouhodobě působící antialergikum (t = 20 hodin) se selektivními H1 antagonistickými vlastnostmi.
Údaje z in vivo (morčata) studií ukazují, že azelastin aplikovaný v terapeuticky relevantních dávkách inhibuje bronchokonstrikci vyvolanou leukotrienem a PAF.
Těmto vlastnostem lze připsat potlačení zánětu respiračního traktu jako základu pro alergické reakce, jak to prokázaly experimenty na zvířatech s azelastini hydrochloridum. Důležitost těchto nálezů z experimentů na zvířatech pro léčbu u člověka není zřejmá.
5.2 Farmakokinetické vlastnosti
Po perorálním podání u zvířat a lidí se azelastini hydrochloridum rychle absorbuje a distribuuje do periferních tkání, zvláště do plic, kůže, svalů, jater a ledvin, ale pouze okrajově do mozku. Byla pozorována kinetika přímo úměrná dávce. Vylučování azelastini hydrochloridum a jeho metabolitů probíhá převážně stolicí (přibližně 75 %) a močí (25 %). Důležité metabolické dráhy zahrnují hydroxylaci kruhu, N-desmethylaci a oxidační otevření azepinového kruhu.
U pacientů s alergickou rinitidou byly pozorovány průměrné plazmatické koncentrace v ustáleném stavu azelastini hydrochloridum dvě hodiny po celkové denní dávce azelastini hydrochloridum 0,56 mg (tj. dvě vstříknutí do každé nosní dírky jednou denně) a dosahovaly přibližně 0,65 ng/ml. Tato plazmatická koncentrace nevedla ke klinicky relevantním systémovým nežádoucím účinkům. Díky přímé úměrnosti dávky se očekává zvýšená plazmatická koncentrace se zvýšením denní dávky.
5.3 Předklinické údaje vztahující se k bezpečnosti
Po opakovaném perorálním podání azelastini hydrochloridum potkanům a psům byly pozorovány první toxické příznaky u dávek, které 75krát převyšovaly maximální denní dávku doporučenou u člověka, což naznačuje, že mají malou důležitost pro klinické použití.
U potkanů se prokázalo, že hlavními cílovými orgány jsou zejména játra (zvýšení aktivity enzymu v séru u AST, ALT a ALP, zvýšení hmotnosti orgánu, hypertrofie buněk a infiltrace tuku) a ledviny (zvýšení hladiny močovinového dusíku a objemu moči, zvýšená eliminace sodíku, draslíku a chloridu, zvýšená hmotnost orgánu). Účinky byly pozorovány pouze po dávkách rovnajících se 200násobnému zvýšení normální perorální denní dávky u člověka.
U mladých a dospělých zvířat nebyly pozorovány žádné toxické účinky při dávkách odpovídajících nejméně 30násobku maximální denní dávky doporučené u člověka.
Intranasální aplikace vysokých dávek azelastini hydrochloridu nosního spreje u potkanů (přibližně 130násobek doporučené intranasální dávky u člověka, vztaženo na tělesnou hmotnost) a psů (přibližně 25násobek doporučené intranasální dávky u člověka, vztaženo na tělesnou hmotnost) po dobu 6 měsíců nevedlo k žádným lokálním nebo orgánově specifickým toxickým účinkům.
Senzibilizační potenciál:
Azelastini hydrochloridum nevykazoval žádný senzibilizační potenciál u morčat.
Mutagenita/kancerogenita:
Azelastini hydrochloridum neprokázal mutagenní potenciál při testech in vitro a in vivo ani žádný karcinogenní potenciál u potkanů a myší.
Embryotoxicita/teratogenita:
V experimentech na zvířatech azelastini hydrochloridum procházel placentou a malá množství byla nalezena v mateřském mléce. Studie embryotoxicity po perorálním podání u potkanů, myší a králíku naznačovaly teratogenní účinky pouze u myší a pozorované účinky se vyskytovaly pouze při toxických dávkách pro samice (68,6 mg/kg/den). Nejnižší embryotoxická perorální dávka byla 30 mg/kg/den u všech tří druhů. Poruchy plodnosti byly pozorovány u samic potkanů při perorálních dávkách vyšších než 3 mg/kg/den.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
dihydrát dinatrium-edetátu hypromelóza
dodekahydrát hydrogenfosforečnanu sodného bezvodá kyselina citronová
chlorid sodný čištěná voda.
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po prvním otevření obalu: nepoužívejte AZELASTIN-POS nosní sprej déle než 6 měsíců po prvním otevření.
6.4 Zvláštní opatření pro uchovávání
Chraňte před chladem nebo mrazem. Neuchovávejte při teplotě nad 25 °C.
6.5 Druh obalu a obsah balení
AZELASTIN-POS nosní sprej je plněn do vícedávkového obalu (z vysokohustotního polyethylenu) s dávkovací pumpičkou. Jedna lahvička obsahuje 10 ml roztoku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
URSAPHARM spol. s r.o.,
Kubánské náměstí 1391/11, 100 00 Praha 10, Česká republika
Tel.: +420 295 560 468
e-mail:
8. REGISTRAČNÍ ČÍSLO(A)
69/149/15-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 18.03.2015
Datum posledního prodloužení regstrace: 1.10.2015
Další informace o léčivu AZELASTIN-POS 1 MG/ML NOSNÍ SPREJ, ROZTOK
Jak
se AZELASTIN-POS 1 MG/ML NOSNÍ SPREJ, ROZTOK
podává: nosní podání - nosní sprej, roztok
Výdej
léku: volně prodejné léčivé přípravky
Balení: Lahev
Velikost
balení: 1X10ML
Držitel rozhodnutí o registraci daného léku v České republice:
URSAPHARM spol. s r.o., Praha
E-mail: info@ursapharm.cz
Telefon: +420 295 560 468
