Souhrnné informace o léku - ACTILYSE
1. NÁZEV PŘÍPRAVKU
ACTILYSE 1 mg/ml prášek a rozpouštědlo pro injekční/infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
1 lahvička s práškem obsahuje:
Alteplasum 10 mg (odpovídá 5 800 000 IU) nebo
Alteplasum 20 mg (odpovídá 11 600 000 IU) nebo
Alteplasum 50 mg (odpovídá 29 000 000 IU)
Alteplasa je vyráběna rekombinantní DNA technikou za použití ovariálních buněk čínského křečka. Specifická aktivita alteplasy jako referenčního materiálu je 580 000 IU/mg. Toto bylo potvrzeno srovnáním s dalším mezinárodním WHO standardem pro t-PA. Specifikace udává specifickou aktivitu alteplasy 522 000 – 696 000 IU/mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční/infuzní roztok
Popis přípravku: bílý až bledě žlutý lyofilizát a čiré bezbarvé rozpouštědlo. Připravený roztok je čirý bezbarvý až bledě žlutý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Trombolytická léčba akutního infarktu myokardu
-
– 90 minutový (zrychlený) dávkovací režim (viz bod 4.2) u pacientů, kde lze léčbu zahájit do
-
– 3 hodiny trvající dávkovací režim (viz bod 4.2) u pacientů, kde lze léčbu zahájit mezi
6–12 hodinami od začátku vzniku příznaků za předpokladu, že je diagnóza zjevně potvrzena. Bylo prokázáno, že Actilyse snižuje 30denní mortalitu u pacientů po akutním infarktu myokardu.
Trombolytická léčba akutní masivní plicní embolie doprovázené hemodynamickou nestabilitou Pokud to stav dovoluje, je vždy vhodné diagnózu ověřit objektivními metodami, tj. angiografií nebo neinvazivními metodami, např. plicní scintigrafií. Nebyl prokázán pozitivní vliv na mortalitu či pozdní morbiditu způsobenou plicní embolií.
Fibrinolytická léčba akutních ischemických cévních mozkových příhod
Léčba musí být zahájena co nejdříve během 4,5 hodiny od vzniku příznaků mozkové příhody a po vyloučení intrakraniálního krvácení vhodnou zobrazovací technikou (například počítačovou tomografií hlavy nebo jinou diagnostickou zobrazovací technikou citlivou na přítomnost krvácení). Efekt léčby je závislý na čase; z tohoto důvodu časnější léčba zvyšuje pravděpodobnost příznivého výsledku léčby.
4.2 Dávkování a způsob podání
Dávkování
Přípravek Actilyse by měl být podán co nejdříve od vzniku příznaků. Je třeba dodržovat následující pokyny pro dávkování.
Akutní infarkt myokardu
a) 90 minutový (zrychlený) dávkovací režim pro pacienty s akutním infarktem myokardu, u nichž lze léčbu zahájit do 6 hodin od vzniku prvních příznaků:
| Koncentrace alteplasy | ||
| 1 mg/ml | 2 mg/ml | |
| ml | ml | |
| 15 mg jako intravenózní bolus | 15 | 7,5 |
| 50 mg v infuzi během 30 minut | 50 | 25 |
| následná infuze 35 mg během 60 minut do maximální dávky 100 mg | 35 | 17,5 |
U osob s tělesnou hmotností (t.h.) do 65 kg musí být dávka upravena dle následující tabulky:
| Koncentrace alteplasy | ||
| 1 mg/ml | 2 mg/ml | |
| ml | ml | |
| 15 mg jako intravenózní bolus | 15 | 7,5 |
| ml/kg t.h. | ml/kg t.h. | |
| 0,75 mg/kg t. h. v infuzi během 30 minut (maximálně 50 mg) | 0,75 | 0,375 |
| následná infuze 0,5 mg/kg t. h. během 60 minut (maximálně 35 mg) | 0,5 | 0,25 |
b) dávkovací režim trvající 3 hodiny pro pacienty, u nichž lze léčbu zahájit za 6 – 12 hodin od vzniku příznaků:
| Koncentrace alteplasy | ||
| 1 mg/ml | 2 mg/ml | |
| ml | ml | |
| 10 mg jako intravenózní bolus | 10 | 5 |
| 50 mg v infuzi během první hodiny | 50 | 25 |
| ml/30 min | ml/30 min | |
| následná infuze 10 mg během dalších 30 minut až do celkové maximální dávky 100 mg během 3 hodin | 10 | 5 |
U osob s tělesnou hmotností do 65 kg nesmí celková dávka přesáhnout 1,5 mg/kg.
Maximální dávka alteplasy je 100 mg.
Pomocná terapie: Pomocná antitrombotická terapie je doporučena podle současných mezinárodních směrnic u pacientů s infarktem myokardu s elevacemi S-T.
Plicní embolie
Celková dávka 100 mg by měla být podána během 2 hodin. Nejlepší výsledky jsou dosahovány při následujícím režimu dávkování:
| Koncentrace alteplasy | ||
| 1 mg/ml | 2 mg/ml | |
| ml | ml | |
| 10 mg jako intravenózní bolus během 1 – 2 minut | 10 | 5 |
| následně 90 mg v nitrožilní infuzi během 2 hodin | 90 | 45 |
U osob s tělesnou hmotností do 65 kg nesmí celková dávka přesáhnout 1,5 mg/kg.
Pomocná terapie: Podávání heparinu je zahajováno (nebo opakovaně zahajováno), dosáhnou-li hodnoty aPTT po aplikaci přípravku Actilyse méně než dvojnásobku horní hranice normálu. Infuze musí být upravena podle výsledků aPTT mezi 50 – 70 sekundami (1,5 – 2,5 násobek výchozí hranice).
Akutní ischemická cévní mozková příhoda (dále CMP)
Léčba musí být prováděna pouze pod dohledem lékaře, který byl vyškolen a má zkušenosti v neurovaskulární péči, viz body 4.3 a 4.4.
Doporučená dávka je 0,9 mg alteplasy/kg tělesné hmotnosti (max. 90 mg) podaná v intravenózní infuzi během 60 minut, přičemž 10 % celkového množství je podáno jako iniciální intravenózní bolus.
Léčbu přípravkem Actilyse je nutno zahájit co nejdříve během 4,5 hodiny od vzniku příznaků. Po 4,5 hodinách od vzniku příznaků cévní mozkové příhody je poměr přínosu a rizika spojený s podáváním přípravku Actilyse negativní a z tohoto důvodu nesmí být přípravek podáván (viz bod 5.1).
Pomocná terapie: Bezpečnost a účinnost tohoto režimu se současným podáním heparinu a kyseliny acetylsalicylové během prvních 24 hodin po vzniku prvních příznaků nebyla dostatečně hodnocena. V průběhu prvních 24 hodin po skončení léčby přípravkem Actilyse nesmí být podána kyselina acetylsalicylová nebo intravenózně heparin. Pokud je podání heparinu nutné v jiné indikaci (např. jako prevence hluboké žilní trombózy), nesmí jeho dávka, podávaná subkutánně, překročit 10 000 IU/den.
Způsob podání
Připravený roztok má být podáván intravenózně. Další praktické informace k přípravě roztoku a jeho podávání jsou uvedeny v bodě 6.6.
Pediatrická populace
Bezpečnost a účinnost přípravku Actilyse nebyla u dětí a dospívajících studována. Podávání přípravku Actilyse je při léčbě akutních cévních mozkových příhod u dětí a dospívajících kontraindikováno (viz bod 4.3).
4.3 Kontraindikace
Obecně u všech indikací nesmí být přípravek Actilyse podáván pacientům se známou hypersenzitivitou na léčivou látku alteplasu, gentamicin (stopový zbytek z výrobního procesu) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Další kontraindikace u akutního infarktu myokardu, akutní plicní embolie a akutní ischemické CMP:
Přípravek Actilyse je kontraindikován v případech vysokého rizika krvácení jako je:
■
■
■
významná porucha krvácení v současnosti či posledních 6 měsících známá hemoragická diatéza
pacienti užívající perorální antikoagulační léčbu, např. warfarin sodný (viz bod 4.4), jejíž účinek je klinicky významný manifestní nebo nedávné silné nebo nebezpečné krvácení
■
■
■
■
v minulosti prodělané intrakraniální krvácení nebo při podezření na něj podezření na subarachnoidální krvácení nebo stavy po subarachnoidálním krvácení z aneurysmatu postižení CNS v anamnéze (např. tumor, aneurysma, chirurgický intrakraniální nebo spinální výkon) nedávná (méně než 10 dnů) traumatická zevní srdeční masáž, porod, nedávná punkce nekomprimovatelných cév (např. punkce v. subclavia nebo v. jugularis) těžká nekontrolovaná arteriální hypertenze bakteriální endokarditida, perikarditida akutní pankreatitida prokázaná gastroduodenální vředová choroba v posledních 3 měsících, jícnové varixy, tepenná aneurysmata, tepenné/žilní malformace nádor se zvýšeným rizikem krvácení závažné jaterní onemocnění včetně jaterního selhání, cirhózy, portální hypertenze (jícnové varixy) a aktivní hepatitidy velký chirurgický výkon nebo významné trauma v posledních 3 měsících.
Další kontraindikace u akutního infarktu myokardu:
známá anamnéza jakékoliv cévní mozkové příhody na podkladě krvácení nebo cévní mozkové příhody neznámého původu
známá anamnéza ischemické cévní mozkové příhody nebo tranzitorní ischemické ataky (TIA)
v předchozích 6 měsících, vyjma současné akutní ischemické cévní mozkové příhody v průběhu posledních 4,5 hodin.
Další kontraindikace u akutní plicní embolie:
známá anamnéza jakékoliv cévní mozkové příhody na podkladě krvácení nebo cévní mozkové příhody neznámého původu
známá anamnéza ischemické cévní mozkové příhody nebo tranzitorní ischemické ataky (TIA)
v předchozích 6 měsících, vyjma současné akutní ischemické cévní mozkové příhody v průběhu posledních 4,5 hodin.
Další kontraindikace u akutní ischemické CMP:
-
■ příznaky ischemické ataky, které započaly před více než 4,5 hodinami před začátkem infuze nebo jestliže není doba nástupu příznaků známa a mohla by být potenciálně před více než 4,5 hodinami (viz bod 5.1).
-
■ neurologický deficit menšího rozsahu nebo rychle se zlepšující příznaky před začátkem infuze
-
■ závažná CMP vyhodnocená klinicky (např. NIHSS>25) a/nebo pomocí příslušné zobrazovací techniky
-
■ epileptický záchvat na počátku CMP
-
■ prokázané intrakraniální krvácení na CT snímku
-
■ příznaky naznačující subarachnoidální krvácení, dokonce i v případě, že CT snímek je v normě
-
■ podávání heparinu během předchozích 48 hodin a tromboplastinový čas převyšující horní hranici normálních laboratorních hodnot
-
■ pacienti s předchozí CMP v anamnéze a současně se vyskytující diabetes
-
■ nedávná CMP během posledních 3 měsíců
-
■ počet krevních destiček pod 100 000/mm3
-
■ systolický krevní tlak (TK) >185 nebo diastolický TK >110 mm Hg nebo útočná léčba (intravenózní farmakoterapie) nezbytná k redukci krevního tlaku (TK) k těmto hodnotám
4.4 Zvláštní upozornění a opatření pro použití
V souladu se zamýšleným použitím je třeba pečlivě zvolit vhodnou velikost balení přípravku Actilyse.
Trombolytická/fibrinolytická léčba vyžaduje patřičnou monitoraci. Přípravek Actilyse by měl být podáván pouze lékaři, kteří byli vyškoleni a mají zkušenosti s trombolytickou léčbou a s vybavením potřebným k monitoraci. Je doporučeno, aby při podávání Actilyse bylo vždy k dispozici standardní resuscitační vybavení a léky.
Hypersenzitivita
Imunitně zprostředkované hypersenzitivní reakce spojené s podáváním Actilyse mohou být způsobeny léčivou látkou alteplasa, gentamicinem (stopový zbytek z výrobního procesu), kteroukoli pomocnou látkou nebo zátkou skleněné lahvičky s práškem Actilyse, která obsahuje přírodní kaučuk (derivát latexu). Po léčbě nebyla pozorována žádná trvalá tvorba protilátek proti molekule rekombinantního humánního tkáňového aktivátoru plazminogenu. Neexistuje žádná systémová zkušenost s opakovaným podáváním Actilyse.
Existuje také riziko hypersenzitivních reakcí zprostředkovaných neimunologickým mechanismem.
Angioedém představuje nejčastější hypersenzitivní reakci hlášenou u přípravku Actilyse. Toto riziko může být zvýšeno v indikaci akutní ischemické cévní mozkové příhody a/nebo při současné léčbě ACE inhibitory (viz bod 4.5). Pacienti léčení v jakékoli schválené indikaci musí být monitorováni z hlediska výskytu angioedému během infuze a po dobu až 24 hodin po infuzi.
Pokud se vyskytne závažná hypersenzitivní reakce (např. angioedém), infuze musí být přerušena a musí být okamžitě zahájena vhodná léčba. Ta může zahrnovat intubaci.
Krvácení
V případě nebezpečného krvácení, zejména do mozku, musí být fibrinolytikum vysazeno. Vzhledem ke krátkému poločasu a minimálnímu účinku na systémové koagulační faktory se ale obecně nevyžaduje náhrada koagulačních faktorů. Krvácení lze ve většině případů léčit vysazením trombolytik a antikoagulačních přípravků, náhradou objemu a manuálním stlačením problémové cévy. U pacientů, kteří dostali během 4 hodin před začátkem krvácení heparin, by měl být zvážen protamin. U malého počtu pacientů, u kterých nebudou tato konzervativní opatření účinná, lze indikovat uvážlivé použití transfuzních produktů. Doporučuje se zvážit transfuzi kryoprecipitátu, čerstvě zmražené plazmy a krevních destiček; po každém podání má následovat kontrolní klinické a laboratorní vyšetření. V případě infuze kryoprecipitátu je žádoucí cílová koncentrace fibrinogenu 1 g/l. Poslední možnost představují antifibrinolytika.
Nebezpečí nitrolebního krvácení je zvýšeno u starších osob, tudíž je nutno u těchto pacientů pečlivě zhodnotit terapeutický prospěch ve srovnání s možným rizikem.
Obdobně jako při podávání jiných trombolytik vyžaduje očekávaný terapeutický prospěch ve srovnání s možným rizikem pečlivé zhodnocení zejména v případech:
■
nedávná menší traumata jako biopsie, punkce větších cév, nitrosvalové injekce, srdeční masáže při resuscitaci
okolnosti vedoucí ke zvýšenému riziku krvácení, které nejsou uvedeny v bodě 4.3.
Je třeba se vyhnout použití rigidních katétrů.
Pacienti užívající perorální antikoagulační léčbu:
Použití Actilyse lze zvážit tehdy, když dávkování nebo doba od posledního podání antikoagulační léčby činí reziduální účinnost nepravděpodobnou, což potvrdí vhodné testy antikoagulační aktivity pro daný přípravek či přípravky, kterých se to týká, které neprokážou žádný klinicky významný vliv na koagulační systém (například pro antagonisty vitamínu K je INR < 1,3, nebo jiné odpovídající testy u dalších perorálních antikoagulačních látek jsou v příslušných horních hranicích normy).
Pediatrická populace
V současné době není dostatek zkušeností s podáváním přípravku Actilyse u dětí a dospívajících.
Další zvláštní upozornění a opatření u akutního infarktu myokardu a akutní plicní embolie: Nesmí se podávat dávky převyšující 100 mg alteplasy, protože mohou být spojeny se zvýšeným výskytem intrakraniálního krvácení. Proto musí být věnována zvláštní péče k zajištění, aby byla dávka alteplasy podána v infuzi tak, jak je předepsáno v bodě 4.2.
Očekávaný terapeutický prospěch musí být zvážen ve srovnání s možným rizikem, zvláště v případech pacientů se systolickým krevním tlakem > 160 mm Hg.
Antagonisté GPIIb/IIIa:
Současné podávání antagonistů GPIIb/IIIa zvyšuje riziko krvácení.
Další zvláštní upozornění a opatření u akutní ischemické CMP:
Zvláštní upozornění pro použití:
Léčba musí být prováděna pouze pod dohledem lékaře, který byl vyškolen a má zkušenosti v neurovaskulární péči. Vhodnou možností ověření správnosti indikované léčby může být dálková diagnostika (viz bod 4.1).
Zvláštní upozornění / podmínky v případech se sníženým poměrem přínos/riziko:
Ve srovnání s ostatními indikacemi mají pacienti s CMP léčení přípravkem Actilyse značně zvýšené riziko intrakraniálního krvácení, protože krvácení se vyskytuje převážně v infarzované oblasti. Toto platí zejména v následujících případech:
■ všechny situace, které jsou zmíněny v bodě 4.3 a obecně všechny stavy s vysokým rizikem krvácení
malá, asymptomatická aneurysmata mozkových cév
při pozdějším času zahájení léčby od vzniku příznaků cévní mozkové příhody se čistý klinický přínos snižuje a může být spojen s vyšším rizikem intrakraniálního krvácení a úmrtí ve srovnání s pacienty léčenými časněji. Proto se nesmí podávání přípravku Actilyse odkládat.
u pacientů předem léčených kyselinou acetylsalicylovou může existovat vyšší riziko intrakraniálního krvácení, zvláště pokud je v léčbě přípravkem Actilyse časová prodleva.
Monitorování krevního tlaku (TK) během léčebné aplikace a až 24 hodin dále poté se zdá být oprávněné; intravenózní antihypertenzní léčba je také doporučena pro systolický TK > 180 mm Hg nebo diastolický TK > 105 mm Hg.
Terapeutický přínos je snížen u pacientů s předchozí CMP nebo se známým nekontrolovaným diabetem, tudíž poměr mezi terapeutickým přínosem a možným rizikem je méně příznivý, avšak přesto je pro tyto pacienty pozitivní.
U pacientů s velmi mírnou CMP riziko převažuje očekávaný přínos (viz bod 4.3).
Pacienti s velmi těžkou CMP mají vyšší riziko intrakraniálního krvácení a úmrtí a neměli by být léčeni (viz bod 4.3).
U pacientů s rozsáhlým infarktem je vyšší riziko nepřesvědčivého výsledku včetně těžkého krvácení a úmrtí. U takových pacientů by měl být pečlivě zvážen poměr mezi přínosem a možným rizikem.
U pacientů s CMP pravděpodobnost dobrých výsledků klesá se zvyšujícím se věkem, vyšší tíží CMP a zvýšenou hladinou krevní glukózy, zatímco pravděpodobnost těžké invalidity a úmrtí nebo závažného intrakraniálního krvácení se zvyšuje nezávisle na léčbě. Pacienti starší než 80 let, pacienti se závažnou CMP (podle klinického obrazu a/nebo stanovením vhodnými zobrazovacími metodami) a pacienti s výchozí hladinou krevní glukózy < 50 mg/100 ml nebo > 400 mg/100 ml by neměli být přípravkem Actilyse léčeni (viz bod 4.3).
Údaje získané ze studie ECASS III a složené analýzy svědčí o tom, že čistý klinický přínos se snižuje u starších pacientů s narůstajícím věkem v porovnání s mladšími pacienty, a to stejně, jako se jeví s narůstajícím věkem přínos léčby přípravkem Actilyse nižší a riziko mortality vyšší.
Další zvláštní upozornění:
Reperfuze ischemické oblasti může vyvolat otok mozku v infarzované zóně.
Pro zvýšené riziko krvácení by léčba inhibitory agregace krevních destiček neměla být zahájena během prvních 24 hodin následujících po trombolytické léčbě s alteplasou.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí mezi přípravkem Actilyse a běžně podávanými léčivými přípravky u pacientů s akutním infarktem myokardu.
Riziko krvácení může být zvýšeno při současném podávání kumarinových derivátů, perorálních antikoagulancií, inhibitorů agregace krevních destiček, nefrakcionovaného heparinu nebo nízkomolekulárního heparinu a léčivých látek ovlivňujících krevní srážlivost (před nebo během prvních 24 hodin po léčbě přípravkem Actilyse) (viz bod 4.3).
Současná léčba ACE inhibitory může zvyšovat riziko vzniku hypersenzitivní reakce (viz bod 4.4).
Současné podávání antagonistů GPIIb/IIIa zvyšuje riziko krvácení.
4.6 Fertilita, těhotenství a kojení
Těhotenství
O použití přípravku Actilyse v průběhu těhotenství jsou dostupné pouze omezené údaje. Neklinické studie s alteplasou v dávkách vyšších, než jsou dávky u lidí, prokázaly nezralost plodu a/nebo embryotoxicitu následkem známého farmakologického účinku přípravku. Má se za to, že alteplasa není teratogenní (viz bod 5.3).
V případě akutního život ohrožujícího onemocnění je nutné před podáním přípravku zvážení prospěchu a eventuálního rizika.
Kojení
Není známo, zda je alteplasa vylučována do mateřského mléka.
Fertilita
O přípravku Actilyse nejsou k dispozici klinické údaje. Neklinické studie s alteplasou neprokázaly žádný nežádoucí účinek na plodnost (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Není relevantní.
4.8 Nežádoucí účinky
Nejčastějším nežádoucím účinkem při podávání přípravku Actilyse je krvácení vedoucí ke snížení hodnot hematokritu a/nebo hemoglobinu.
Nežádoucí účinky uvedené níže jsou rozdělené podle frekvence výskytu a tříd orgánových systémů. Skupiny četnosti jsou definovány za použití následujícího pravidla: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10 000 až <1/1000); velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
S výjimkou intrakraniálního krvácení jako nežádoucího účinku při indikaci CMP a reperfuzní arytmie při indikaci infarkt myokardu není žádný medicínský důvod předpokládat, že kvalitativní a kvantitativní profil nežádoucích účinků přípravku Actilyse při indikaci plicní embolie a CMP bude rozdílný od profilu při indikaci infarkt myokardu.
Tabulka 1 Nežádoucí účinky u infarktu myokardu, plicní embolie a ischemické cévní mozkové příhody
| Třída orgánových systémů | Nežádoucí účinek |
| Krvácení | |
| velmi časté | intracerebrální krvácení představuje hlavní nežádoucí účinek při léčbě akutní ischemické cévní mozkové příhody (až u 15 % pacientů bez jakéhokoliv kombinovaného významného zvýšení celkové mortality a závažné invalidity, tj. s mRS 5 až 6). krvácení z poškozených krevních cév (jako je hematom) |
| časté | intrakraniální krvácení (jako je krvácení do mozku, hematom mozku, krvácivá cévní mozková příhoda, hemoragická transformace ischemické cévní mozkové příhody, intrakraniální hematom, subarachnoidální krvácení) při léčbě akutního infarktu myokardu a akutní plicní embolie krvácení v hltanu gastrointestinální krvácení (jako je krvácení žaludeční, krvácení ze žaludečního vředu, krvácení z konečníku, hemateméza, meléna, krvácení z úst, krvácení z dásní) ekchymózy urogenitální krvácení (jako je hematurie, krvácení z močových cest) krvácení v místě injekce (krvácení v místě vpichu, hematom v místě zavedení katetru, krvácení v místě zavedení katetru) |
| méně časté | plicní krvácení (jako je hemoptýza, hemothorax, krvácení v respiračním traktu) epistaxe ušní krvácení |
| vzácné | oční krvácení perikardiální krvácení retroperitoneální krvácení (jako je retroperitoneální hematom) |
| není známo | krvácení v parenchymatózních orgánech (jako je jaterní krvácení) |
| Poruchy imunitního systému* | |
| vzácné | hypersenzitivní reakce (např. vyrážka, kopřivka, bronchospasmus, angioedém, hypotenze, šok)* |
| velmi vzácné | závažná anafylaxe |
| Poruchy nervového systému | |
| velmi vzácné | příhody mající vztah k nervovému systému (např. epileptický záchvat, křeče, afázie, porucha řeči, delirium, akutní mozkový |
| syndrom, vzrušení, zmatenost, deprese, psychóza) často spojené se současnými ischemickými nebo hemoragickými cerebrovaskulárními příhodami | |
| Srdeční poruchy | |
| velmi časté | rekurentní ischemie / angína, hypotenze a srdeční selhání / plicní edém |
| časté | kardiogenní šok, zástava srdce a reinfarkt |
| méně časté | reperfuzní arytmie (jako jsou arytmie, extrasystoly, AV blok I. stupně až kompletní AV blok, fibrilace síní / flutter síní, bradykardie, tachykardie, komorové arytmie, komorová tachykardie / fibrilace komor, elektromechanická disociace [EMD]) mitrální regurgitace, plicní embolie, jiná systémová embolie / cerebrální embolie, ventrikulární septální defekt |
| Cévní poruchy | |
| vzácné | embolie, která může vést k odpovídajícím důsledkům v postižených orgánech |
| Gastrointestinální poruchy | |
| vzácné | nauzea |
| není známo | zvracení |
| Vyšetření | |
| méně časté | pokles krevního tlaku |
| není známo | zvýšená tělesná teplota |
| Poranění, otravy a procedurální komplikace | |
| není známo | tuková embolie (embolizace krystalů cholesterolu), která může vést k odpovídajícím důsledkům v postižených orgánech |
| Chirurgické a léčebné postupy | |
| není známo | transfuze krve (nezbytná) |
*Viz body 4.4. a 4.5.
Srdeční poruchy
Jako u jiných trombolytických látek byly hlášeny následující příhody jako následky infarktu myokardu a/nebo trombolytické léčby. Tyto srdeční příhody mohou být život ohrožující a mohou vést k úmrtí.
Výpočet frekvence výskytu
Tento nežádoucí účinek byl pozorován po uvedení přípravku na trh. S 95% jistotou není frekvence výskytu větší než „vzácné“, ale může být nižší. Přesný odhad frekvence výskytu není možný, jelikož se nežádoucí účinek nevyskytl v databázi klinických hodnocení s 8299 pacienty.
Úmrtí a trvalá invalidita byly hlášeny u pacientů, kteří prodělali CMP (včetně intrakraniálního krvácení) a jiné závažné příhody spojené s krvácením.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv
Šrobárova 48
100 41 Praha 10
Webové stránky:
4.9 Předávkování
Přes relativní fibrinovou specificitu přípravku Actilyse může při předávkování docházet ke klinicky signifikantní redukci fibrinogenu a ostatních složek krevní koagulace. Ve většině případů dostačuje vyčkat fyziologické regenerace uvedených faktorů po skončení terapie přípravkem Actilyse. Pokud však dojde k závažnému krvácení, je doporučena infuze čerstvé mražené plazmy a v odůvodněných případech mohou být podána syntetická antifibrinolytika.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antitrombotika, ATC kód: B01AD02
Léčivá látka přípravku Actilyse je alteplasa, což je rekombinantní lidský tkáňový aktivátor plazminogenu, glykoprotein, který aktivuje plazminogen přímo na plazmin. Při nitrožilním podání zůstává v krevním oběhu alteplasa relativně inaktivní. Po její vazbě na fibrin je však aktivována, indukuje konverzi plazminogenu na plazmin, mající za následek rozpuštění fibrinové sraženiny.
Vzhledem ke své relativní specificitě k fibrinu vede alteplasa v dávce 100 mg k mírnému poklesu hladin cirkulujícího fibrinogenu asi na 60 % původních hodnot po 4 hodinách. Tento účinek je reverzibilní, takže po 24 hodinách je hladina fibrinogenu asi 80%. Plazminogen a alfa-2-antiplazmin se snižují asi na 20 %, resp. 35 %, po 4 hodinách a opět vzrůstají na více než 80 % po 24 hodinách. Výrazný a dlouhodobý pokles hladin cirkulujícího fibrinogenu je patrný pouze u minima pacientů.
Ve studii, která zahrnovala více než 40 000 pacientů s akutním infarktem myokardu (GUSTO), vedlo podávání 100 mg alteplasy během 90 minut se současným podáním infuze heparinu intravenózně k nižší mortalitě po 30 dnech (6,3 %) v porovnání s podáním 1,5 milionu IU streptokinázy během 60 minut spolu s heparinem podaným subkutánně nebo intravenózně (7,3 %). Alteplasou léčení pacienti vykazovali vyšší frekvenci průchodnosti cév se vztahem k infarktu po 60 a 90 minutách po trombolýze oproti pacientům léčeným streptokinázou. Po 180 minutách a déle nebyly zaznamenány žádné rozdíly v průchodnosti cév.
30-denní mortalita se snížila v porovnání s pacienty, kteří nepodstoupili trombolytickou léčbu.
Uvolňování alfa-hydroxybutyrát-dehydrogenázy (HBDH) se snížilo. Globální systolická funkce komor stejně jako regionální porucha kinetiky komor byly méně narušeny při srovnání s pacienty, kteří nepodstoupili trombolytickou léčbu.
Infarkt myokardu
Placebem kontrolovaná studie s podáním 100 mg alteplasy během 3 hodin (LATE) ukázala snížení 30-denní mortality ve srovnání s placebem u pacientů léčených během 6–12 hodin od vzniku příznaků. V případech, kdy jsou příznaky infarktu myokardu jasně vyjádřeny, může být stále ještě přínosná i léčba zahájená až za 24 hodin od vzniku příznaků.
Plicní embolie
U pacientů s akutní masivní plicní embolií s hemodynamickou nestabilitou vede trombolytická léčba přípravkem Actilyse k rychlému zmenšení velikosti trombu a ke snížení tlaku v arteria pulmonalis. Údaje týkající se mortality nejsou dostupné.
Pacienti s akutní ischemickou cévní mozkovou příhodou
Ve dvou studiích v USA (NINDS A/B) měl významně vyšší podíl pacientů příznivý výstup při podávání alteplasy ve srovnání s placebem (žádná nebo minimální invalidita). Tato zjištění byla potvrzena ve studii ECASS III (viz odstavec níže), poté, co mezitím dvě evropské studie a další studie v USA selhaly při poskytnutí příslušného důkazu při nastaveních, která v podstatě nevyhovují aktuální informaci o přípravku v EU.
Studie ECASS III byla placebem kontrolovaná dvojitě zaslepená studie provedená v Evropě u pacientů s akutní cévní mozkovou příhodou v časovém okně 3 až 4,5 hodiny. Léčebné podání ve studii ECASS III bylo v souladu s evropským souhrnem údajů o přípravku (SmPC) přípravku Actilyse pokud jde o jeho indikaci u cévní mozkové příhody s výjimkou horního limitu času léčebného okna, tj. 4,5 hodiny. Primárním cílem byla invalidita za 90 dní rozdělená do dvou skupin – na skupinu s příznivým výstupem (na upravené stupnici Rankin [mRS] stupeň 0 až 1) a na skupinu s nepříznivým výstupem (mRS stupeň 2 až 6). Celkem bylo randomizováno 821 pacientů (418 alteplasa/403 placebo). Více pacientů dosáhlo příznivého výstupu s alteplasou (52,4 %) oproti placebu (45,2 %; poměr šancí (OR = odds ratio) 1,34; 95% interval spolehlivosti (CI) 1,02 – 1,76; p=0,038). Výskyt jakéhokoli intrakraniálního krvácení/symptomatického intrakraniálního krvácení byl vyšší u alteplasy oproti placebu (jakékoli intrakraniální krvácení 27,0 % proti 17,6 %, p=0,0012; symptomatické intrakraniální krvácení podle definice studie ECASS III 2,4 % oproti 0,2 %, p=0,008). Mortalita byla nízká a nelišila se významně mezi alteplasou (7,7 %) a placebem (8,4 %; p=0,681). Výsledky podskupin studie ECASS III potvrzují, že delší čas od vzniku cévní mozkové příhody do zahájení léčby (OTT) je spojen s narůstajícím rizikem mortality a symptomatického intrakraniálního krvácení. Výsledky studie ECASS III ukazují pozitivní čistý klinický přínos přípravku Actilyse během časového okna 3 až 4,5 hodiny, zatímco složené údaje demonstrují, že čistý klinický přínos přestává být pro alteplasu příznivý v časovém okně delším než 4,5 hodiny.
Bezpečnost a účinnost léčby přípravkem Actilyse u akutní ischemické cévní mozkové příhody až do 4,5 hodiny času od vzniku cévní mozkové příhody do zahájení léčby (OTT) byl hodnocen
v pokračujícím registru (SITS-ISTR: The Safe Implementation of Thrombolysis in Stroke registry = Bezpečné provedení trombolýzy v registru cévních mozkových příhod). V této observační studii byly srovnávány výstupní údaje o bezpečnosti 21 566 pacientů léčených v časovém okně 0 až 3 hodiny s údaji 2376 pacientů léčenými mezi 3 a 4,5 hodinami po začátku akutní ischemické cévní mozkové příhody. Bylo zjištěno, že výskyt symptomatického intrakraniálního krvácení (podle definice SITS-MOST), je vyšší v časovém okně od 3 do 4,5 hodiny (2,2 %) ve srovnání s časovým oknem do 3 hodin (1,7 %). Frekvence mortality za 3 měsíce byly podobné při srovnání časového okna 3 až 4,5 hodiny (12,0 %) s časovým oknem 0 až 3,0 hodiny (12,3 %), s poměrem šancí (OR) bez adjustace 0,97 (95% CI: 0,84–1,13, p=0,70), a poměrem šancí (OR) po adjustaci 1,26 (95% CI: 1,07–1,49, p=0,005). Observační údaje SITS podporují důkaz z klinické studie, že čas od vzniku cévní mozkové příhody do zahájení léčby (OTT) je při léčbě akutní cévní mozkové příhody alteplasou důležitým faktorem předurčujícím výstup.
5.2 Farmakokinetické vlastnosti
Alteplasa je rychle odstraňována z cirkulující krve a metabolizována hlavně v játrech (plazmatická clearance 550–680 ml/min.). Plazmatický poločas t1/2 alfa je 4–5 minut. To znamená, že po 20 minutách je v plazmě přítomno méně než 10% počátečního množství přípravku. Pro zbylé množství přípravku v těžce dostupném kompartmentu, byl naměřen beta-poločas okolo 40 minut.
5.3 Předklinické údaje vztahující se k bezpečnosti
V subchronických studiích toxicity provedených u laboratorních potkanů a opic druhu kosman nebyly zjištěny neočekávané nežádoucí účinky. V testech mutagenity nebyl shledán žádný mutagenní potenciál.
U březích zvířat nebyly po nitrožilní infuzi farmakologicky účinných dávek pozorovány teratogenní účinky. Embryotoxicita u králíků (letalita embryí, retardace růstu) byla indukována podáním dávky přesahující 3 mg/kg/den. U potkanů nebyly poruchy peri- a postnatálního vývoje a fertility pozorovány při podání dávek až do 10 mg/kg/den.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Arginin, koncentrovaná kyselina fosforečná, polysorbát 80
Rozpouštědlo:
Voda pro injekci
6.2 Inkompatibility
Přípravený roztok může být dále ředěn fyziologickým roztokem (sterilním roztokem chloridu sodného na injekci o koncentraci 9 mg/ml = 0,9 %) až na minimální koncentraci 0,2 mg alteplasy na 1 ml.
K dodatečnému naředění se nedoporučuje používat vodu pro injekci nebo obecně infuzní roztoky cukrů, například glukosy, a to z důvodu zvýšené tvorby zákalu v připravovaném roztoku.
Přípravek Actilyse se nesmí mísit s jinými léky ve stejné infuzní láhvi a nesmí být s jinými léky podáván stejným katetrem (ani s heparinem).
6.3 Doba použitelnosti
Neotevřené lahvičky
3 roky
Rekonstituovaný roztok
Chemická a fyzikální stabilita připraveného roztoku byla prokázána po dobu 24 hodin při teplotě 2 – 8°C, a po dobu 8 hodin při teplotě 25°C.
Z mikrobiologického hlediska je třeba roztok použít bezprostředně po přípravě. Pokud není použit okamžitě, pak jsou podmínky uchovávání i doba uchovávání připraveného roztoku na zodpovědnosti uživatele a za normálních okolností nesmí přesáhnout dobu 24 hodin při teplotě 2 – 8°C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Uchovávejte při teplotě do 25°C.
Podmínky uchovávání přípravku po naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Prášek:
10 ml, 20 ml nebo 50 ml sterilní skleněné lahvičky se zátkou ze silikonizované šedé butylové pryže, s hliníkovým uzávěrem a krytem.
Rozpouštědlo:
Voda pro injekci je plněna do 10, 20 nebo 50 ml skleněných lahviček, v závislosti na velikosti lahviček s práškem. Lahvičky s vodou pro injekci jsou uzavřeny pryžovou zátkou, hliníkovým uzávěrem a krytem.
Přeplňovací kanyly (pouze u velikostí balení 20 a 50 mg)
Velikosti balení:
10 mg:
1 lahvička s 467 mg prášku pro přípravu injekčního a infuzního roztoku
1 lahvička s 10 ml vody pro injekci
20 mg:
1 lahvička s 933 mg prášku pro přípravu injekčního a infuzního roztoku
1 lahvička s 20 ml vody pro injekci
1 přeplňovací kanyla
50 mg:
1 lahvička s 2333 mg prášku pro přípravu injekčního a infuzního roztoku
1 lahvička s 50 ml vody pro injekci
1 přeplňovací kanyla
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Pro rekonstituci na konečnou koncentraci 1 mg alteplasy na 1 ml se převede celý objem rozpouštědla do lahvičky obsahující prášek přípravku Actilyse. Pro tento účel je do balení po 20 mg a 50 mg vložena přeplňovací kanyla. Pro velikost balení 10 mg se musí použít injekční stříkačka.
Pro rekonstituci na konečnou koncentraci 2 mg alteplasy na 1 ml musí být použit pouze poloviční objem rozpouštědla (jak uvádí tabulka níže). V těchto případech musí být vždy použita injekční stříkačka pro převedení potřebného objemu rozpouštědla do lahvičky obsahující prášek přípravku Actilyse.
Za aseptických podmínek se obsah lahvičky Actilyse (10 mg nebo 20 mg nebo 50 mg) rozpustí ve vodě pro injekci dle následující tabulky tak, aby bylo dosaženo výsledné koncentrace 1 mg alteplasy na 1 ml nebo 2 mg alteplasy na 1 ml:
| Actilyse suchý prášek | 10 mg | 20 mg | 50 mg |
| (a) Množství vody pro injekci, které je nutno | 10 ml | 20 ml | 50 ml |
| přidat k suchému prášku Výsledná koncentrace: | 1 mg alteplasy/ml | 1 mg alteplasy/ml | 1 mg alteplasy/ml |
| (b) Množství vody pro injekci, které je nutno přidat k suchému prášku | 5 ml | 10 ml | 25 ml |
| Výsledná koncentrace: | 2 mg alteplasy/ml | 2 mg alteplasy/ml | 2 mg alteplasy/ml |
Připravený roztok má být poté podáván intravenózně. Připravený roztok o koncentraci 1 mg/ml lze dále naředit fyziologickým roztokem (sterilním roztokem chloridu sodného pro injekci o koncentraci 9 mg/ml = 0,9 %) až na minimální koncentraci 0,2 mg/ml. Připravený roztok o koncentraci 1 mg/ml se nedoporučuje dále ředit sterilní vodou pro injekci nebo obecně používáním infuzních roztoků cukrů, například glukosy. Actilyse se nesmí mísit s jinými léky ve stejné infuzní láhvi a nesmí být s jinými léky podávána stejným katetrem (ani s heparinem).
Inkompability – viz bod 6.2.
Připravený roztok je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Pokyny pro rekonstituci přípravku Actilyse
| 1. | Roztok připravujte bezprostředně před podáním. | |
| 2. | Odstraňte ochranné kryty z obou lahviček tak, že je odklopíte palcem ruky. V jedné lahvičce je sterilní voda pro injekci, v druhé suchý prášek Actilyse. | ( ■ <5^ Boehli»R" \ ( 1 i f'Š\ Bochtirt*' \ ZXI^lfr lngellieim__^A. InKclbt^^-Ay |
| 3. | Dezinfikujte pryžovou zátku obou lahviček pomocí tampónu s alkoholem. |
| 4. | Vyjměte přeplňovací kanylu* z jejího obalu. Kanylu není třeba dezinfikovat či sterilizovat, je sterilní. Sejměte z kanyly jeden z jejích dvou krytů. | 1 o |
| 5. | Postavte lahvičku se sterilní vodou pro injekci na stabilní vodorovnou plochu. Vertikálně ve směru shora dolů zasuňte přeplňovací kanylu do lahvičky středem pryžové zátky. K zasunutí kanyly použijte jemný, ale pevný tlak, kanylou přitom neotáčejte. | Sterilní voda na injekci í ů < l |
| 6. | Jednou rukou uchopte pevně lahvičku se sterilní vodou pro injekci spolu s přeplňovací kanylou. Lahvičku uchopte do dlaně, dvě postranní křidélka kanyly přidržte prsty. | |
| Druhou rukou odstraňte zbývající kryt z přeplňovací kanyly. | Tu |
7.
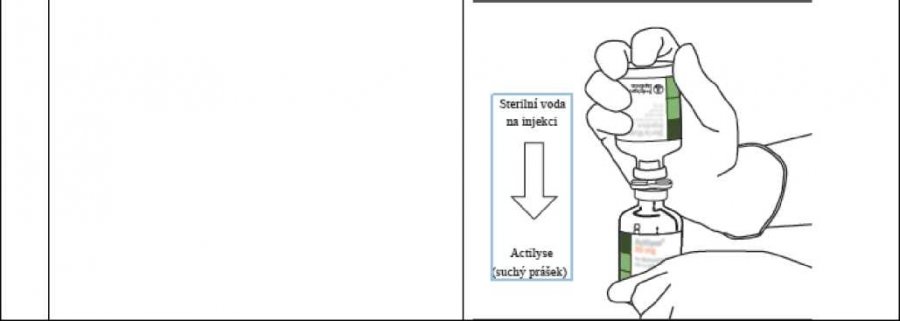
Lahvičku se sterilní vodou pro injekci držte jednou rukou pevně v dlani, dvě postranní křidélka kanyly přidržujte prsty.
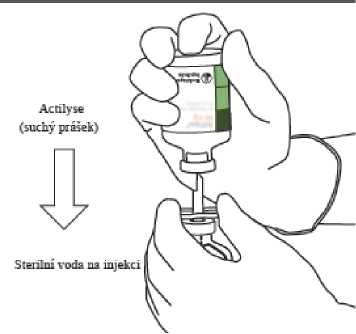
Umístěte lahvičku se suchým práškem Actilyse nad přeplňovací kanylu tak, aby špička kanyly byla přesně ve středu pryžové zátky lahvičky se suchým práškem Actilyse.
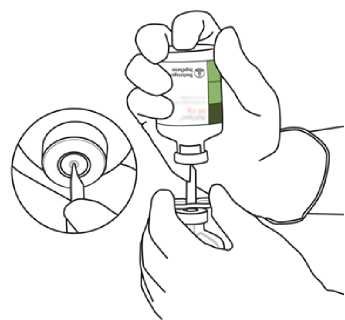
Lahvičku se suchým práškem Actilyse tlačte vertikálně ve směru shora dolů proti špičce přeplňovací kanyly, čímž proniknete pryžovou zátkou lahvičky. Vyvíjejte jemný, ale pevný tlak, aniž byste kanylou či lahvičkou otáčeli.
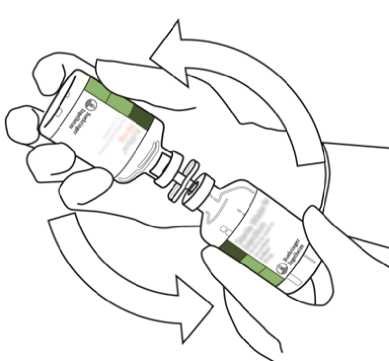
8.
Přetočte obě lahvičky tak, aby se obsah lahvičky s vodou pro injekci zcela přelil do lahvičky se suchým práškem Actilyse.

| 9. | Odstraňte prázdnou lahvičku na vodu pro injekci spolu s přeplňovací kanylou. Můžete je vyhodit do odpadu. | 1 |
| 10. | Vezměte lahvičku s připraveným roztokem Actilyse a jemně jí zakružte, aby se veškerý zbývající prášek rozpustil. Lahvičku ale neprotřepávejte, protože to by vedlo ke vzniku pěny. Pokud se v roztoku objevily bubliny, nechte jej po dobu několika minut nerušeně odstát. Bubliny vymizí. | |
| 11. | Připravený roztok má koncentraci 1 mg/ml. Je čirý, bezbarvý až bledě žlutý. Nesmí obsahovat žádné pevné částice. | |
| 12. | Pomocí jehly a injekční stříkačky odměřte požadované množství roztoku. K zabránění úniku roztoku zavádějte jehlu mimo místo, kudy do pryžové zátky dříve pronikla přeplňovací kanyla. | |
| 13. | Použijte okamžitě. Nespotřebovaný roztok znehodnoťte. |
| (*po | kud je přeplňovací kanyla součástí balení přípravku. Přípravu roztoku lze též provést pomocí |
injekční stříkačky a jehly.)
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Boehringer Ingelheim International GmbH
Ingelheim am Rhein, Německo
8. REGISTRAČNÍ ČÍSLO
16/414/92-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 1.7.1992
Datum posledního prodloužení registrace: 21.11.2012
Další informace o léčivu ACTILYSE
Jak
se ACTILYSE
podává: intravenózní podání - prášek a rozpouštědlo pro injekční/infuzní roztok
Výdej
léku: na lékařský předpis
Balení: Injekční lahvička
Velikost
balení: 1+1X10ML
Držitel rozhodnutí o registraci daného léku v České republice:
Boehringer Ingelheim International GmbH, Ingelheim am Rhein
E-mail: infocz@boehringer-ingelheim.com